Newsletter ZKS
Regulatory Affairs
Klicken für Zugang zur Präsentation der EMA
01.10.2025
Neue ICH GCP E6 (R3) in Kraft getreten
Mit Wirkung vom 23. Juli 2025 tritt die überarbeitete Fassung der internationalen Leitlinie für Gute Klinische Praxis (GCP), ICH GCP E6 (R3), in Kraft.
Die Revision reagiert auf den technologischen und methodischen Fortschritt in der klinischen Forschung, einschließlich digitaler und dezentraler Ansätze.
Wesentliche Neuerungen im Überblick:
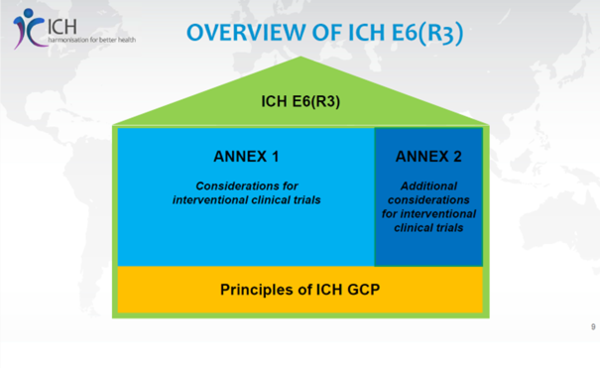
- Modernisierte Struktur: Die neue Fassung besteht aus einem Core-Dokument, dem Annex 1 (Interventionelle Studien), und einem zukünftig erscheinenden Annex 2 (dezentrale Studienelemente)
- Quality by Design: Qualitätsaspekte sollen bereits in der Planungsphase von Studien berücksichtigt werden.
- Risikobasiertes Qualitätsmanagement: Fokus auf relevante Risiken für Datenqualität und Patientensicherheit.
- Flexibilität für diverse Studiendesigns: Die Leitlinie bietet mehr Spielraum für klassische, hybride und dezentrale Studien.
- Digitale Technologien: Elektronische Datenerfassung und digitale Kommunikation werden explizit berücksichtigt.
- Data Governance: Ein neuer Abschnitt bekräftigt die zentrale Bedeutung von Datenintegrität, digitalen Systemen und technologiegestützten Prozessen.
Warum ist die Revision wichtig?
Die klinische Forschung hat sich in den letzten Jahren stark verändert – durch Digitalisierung, dezentrale Studien und komplexe Prüfdesigns. Die neue Leitlinie trägt diesen Entwicklungen Rechnung.
Schwerpunkte der Überarbeitung
Im Mittelpunkt der Revision standen die Harmonisierung mit anderen ICH-Leitlinien und die Schließung inhaltlicher Lücken. Der Geltungsbereich bleibt weiterhin auf klinische Prüfungen mit Humanarzneimitteln beschränkt. Gleichzeitig wurden die Grundprinzipien und Ziele der GCP modernisiert. Besonderer Wert wurde auf größtmögliche Flexibilität hinsichtlich verschiedener Studiendesigns, Organisationsformen und technologieoffener Datennutzung gelegt – um die Richtlinie zukunftsfähig zu gestalten.
Neuer Aufbau der ICH GCP E6 (R3)
ICH GCP E6 R3 umfasst das Core-Dokument sowie den Annex 1. Für die Umsetzung galt eine Übergangsfrist bis zum 23. Juli 2025. Der Annex 2 wird voraussichtlich im 3. Quartal 2026 veröffentlicht.
Die Überarbeitung der bisherigen Version E6 (R2), die seit 2017 in Kraft war, wurde vom International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) angestoßen. Ziel war es, neue Prüfkonzepte, digitale Technologien und moderne Datenquellen stärker zu berücksichtigen.
Um den Anforderungen heutiger klinischer Forschung gerecht zu werden und ausreichend Flexibilität für zukünftige technologische Entwicklungen zu schaffen, wurde die Guideline grundlegend neu strukturiert. Anstelle eines einzelnen Dokuments besteht die neue Fassung nun aus einem überarbeiteten Core-Dokument, mehreren Annexen (1 und künftig 2) sowie ergänzenden Appendices.
Annex-Struktur und Studiendesigns
Das Core-Dokument und Annex 1 gelten für konventionelle, interventionelle klinische Studien.
Der zukünftige Annex 2 soll zusätzliche Leitlinien für dezentrale, pragmatische Studiendesigns und solche, die auf Real-World-Daten (RWD) basieren, enthalten.
Die überarbeiteten GCP-Prinzipien in E6 (R3) sind:
zeitlos formuliert,
technologieoffen,
risikobasiert,
und bieten Flexibilität für klassische, hybride und dezentrale Studien.
Besonders hervorgehoben wird die Rolle moderner Technologien, wie digitale Tools, eConsent oder eSource-Systeme.
Der risikobasierte Ansatz für Planung, Durchführung und Überwachung klinischer Studien wird in R3 noch stärker betont als in der Vorgängerversion.
Neu: Abschnitt „Data Governance“
Ein ganz neuer Abschnitt widmet sich dem Thema Data Governance. Er unterstreicht die zentrale Bedeutung von Datenintegrität, digitalen Systemen und technologiegestützten Prozessen in der heutigen klinischen Forschung. Alle Daten – egal ob papierbasiert, elektronisch oder aus Apps und Geräten – müssen vollständig, korrekt, nachvollziehbar und sicher erfasst und gespeichert werden. In diesem Bereich sind innovative Ansätze von Sponsoren, Dienstleister und Studiendurchführenden gefragt, um die Studienlandschaft zukunftsfähig aufzustellen.
Die vollständige Leitlinie ICH GCP E6 (R3) ist hier verfügbar.

01.04.2024
Regulatory affairs
Nach dem Start im September 2022 hat die EMA drei akademische und gemeinnützige Organisationen, die Arzneimittel für neuartige Therapien (ATMP) entwickeln, in ein Pilotprogramm aufgenommen, in dessen Rahmen sie von einer verstärkten Unterstützung durch die EMA profitieren. Potenzielle Forschende können sich bewerben, indem sie das auf der EMA-Website verfügbare Bewerbungsformular ausfüllen oder die EMA über atmppilot@ema.europa.eu kontaktieren, um ihr Interesse an der Teilnahme am Pilotprogramm zu bekunden oder weitere Informationen zu erhalten. Ein aktualisiertes Dokument mit Fragen und Antworten ist ebenfalls verfügbar.
Übergang der klinischen Prüfungen zum neuen EU-System - noch ein Jahr
Alle laufenden klinischen Prüfungen mit Arzneimitteln in der EU müssen bis zum 31. Januar 2025 in das Clinical Trials Information System (CTIS) überführt und damit unter der sogenannten Clinical Trials regulation (CTR, Verordnung über klinische Prüfungen EU-VO 536/2014) weitergeführt werden. Dieses Datum markiert das Ende einer dreijährigen Übergangsfrist, die mit dem Inkrafttreten der CTR n der EU begann.
Verordnung über klinische Prüfungen (EU) - Fragen und Antworten, Version 6.8
Dieses Dokument enthält häufig gestellte Fragen und Antworten zur Umsetzung der Vorschriften über klinische Prüfungen. Aktualisierungen dieses Fragen- und Antwortdokuments werden in der "Expertengruppe für klinische Prüfungen" vorgestellt und diskutiert und spiegeln die Meinung der Gruppe wider. Diese Gruppe wird von der Kommission geleitet und setzt sich aus Vertretern aller EU-Mitgliedstaaten und EWR-Vertragsparteien zusammen.
01.11.2023
EMA: Auslaufen der außergewöhnlichen Flexibilitäten im Zusammenhang mit COVID-19
Die Europäische Arzneimittelagentur (EMA), die Europäische Kommission (EK) und die Leiter der Arzneimittelagenturen (HMA) gaben im Juli 2023 bekannt, dass sie die außerordentliche regulatorische Flexibilität (“extraordinary regulatory flexibilitys“), die während der Covid-19-Pandemie zur Bewältigung der Regulierung und Versorgung von Arzneimittel eingerichtet wurde, auslaufen lassen. Damit folgen die Behörden der WHO, die im Mai 2023 den COVID-19-Gesundheitsnotstand für beendet erklärte.
Q&A: Good Clinical Practice
(D3) Neu im Juni 2023: Welche Überlegungen sind erforderlich, wenn ein direkter Fernzugriff auf identifizierbare persönliche und Gesundheitsdaten in einer klinischen Studie notwendig ist?
Direkter Fernzugriff ist jeglicher Zugriff von einem Zugangspunkt (Ort und/oder Hardware), der nicht unter der Kontrolle und Aufsicht des Prüfers oder der Institution steht. Direkter Fernzugriff beinhaltet zusätzliche Datenverarbeitung.
Überlegungen zu:
- Der direkte Fernzugriff muss in der Patienteninformation klar erläutert werden.
- Klinisches Studienprotokoll muss gemäß Verordnung (EU) Nr. 536/2014 eine Beschreibung darüber enthalten, wie die Vorkehrungen für den Fernzugriff mit den geltenden Datenschutzbestimmungen übereinstimmen.
- Eine Datenschutz-Folgenabschätzung sollte erfolgen.
(B19) Neu im August 2023: Was sind die Erwartungen bezüglich der Verteilung aktualisierter Investigators Brochures (IBs) und aktualisierter Einverständniserklärungen (ICFs) an klinische Prüfstellen bzw. Prüfer?
Eine IB, welche wesentliche Änderungen erhält, sollte unverzüglich nach der Genehmigung an die Prüfstellen verteilt werden. Es ist nicht akzeptabel, auf die Genehmigung für alle betroffenen EU-Mitgliedsstaaten oder weltweit zu warten.
Ebenso sollte eine überarbeitete Patienteninformation, sobald die Genehmigungen vorliegen, an die Prüfstellen verteilt werden. Eine nicht gerechtfertigte erhebliche Verzögerung, die die Rechte der Patienten beeinträchtigt, zwischen der Stellungnahme/Genehmigung und der Verteilung (z.B. mehrere Wochen) gilt als inakzeptabel.
Informed Consent bei pädiatrischen Studien
Das European Network of Paediatric Research der EMA (EnprEMA) hat im Juni 2023 eine Übersicht der Anforderungen von 28 europäischen Ländern des Informed Consent bei pädiatrischen klinischen Studien veröffentlicht.
Der Studienteilnehmer wurde nicht ausreichend informiert bzw. eine Nichtkonformität mit den GCP-Richtlinien liegt vor, wenn:
- ein veraltetes ICF , in dem wichtige neue Informationen fehlen, beim Einschluss vom Teilnehmer unterzeichnet wurde.
- es zu einer Verzögerung bei der Einholung einer erneuten Zustimmung zur Teilnahme an einer Studie auf der Grundlage eines neuen ICF kommt.
NEU: Klinikumsweite Verfahrensanweisung: Patientenaufklärung
Die klinische Prüfung eines Arzneimittels oder eines Medizinproduktes am Menschen sowie sonstige medizinisch-wissenschaftliche Studien („Sonstige“ Studien oder auch Non-AMG/Non-MPDG-Studien) dürfen nur mit Einwilligung nach Aufklärung des Prüfungs- oder Studienteilnehmenden begonnen werden.
Um den Prozess der Patientenaufklärung im Rahmen von klinischen Studien an allen studienführenden Abteilungen des UKF zu harmonisieren, wurde die klinikumsweite Verfahrensanweisung „Patientenaufklärung“ (Dok.-ID 93264) erstellt.
Damit haben Studienleiter*innen, Wissenschaftler*innen und Forschende eine allgemeingültige Verfahrensanweisung an der Hand, die die Prozesse rund um die Aufklärung und Einholung der Einwilligungserklärung von potentiellen Studienteilnehmenden für das UKF regelt.
Die klinikumsweite Verfahrensanweisung für die Patientenaufklärung im Rahmen von klinischen Studien ist gültig seit dem 7.11.2023 und hier zu finden: roxTra Dok.-ID 93264 .
Arzneimittelgesetz
Seit 31.01.2022 gilt die neue EU-Verordnung 536/2014 (Clinical Trial Regulation, CTR) für AMG Studien. Alle jetzt neu einzureichenden AMG Studien müssen zentral über das neue EU Portal CTIS eingereicht werden.
Zugehörige klinikumsweite Arbeitsanweisungen und Checklisten sind in roXtra hinterlegt:
Dok.-ID 81563 CTIS UKF Organisation ID und Location IDs der Kliniken, Institute und Einrichtungen
Dok.-ID 80584 Anlegen einer neuen AMG UKF IIT im CTIS Portal und Benennung CT-Administrator*in
Dok.-ID 79768 UKF IIT Benennung eines CTIS-CT-Administrators
Clinical Updates
Die EMA hat die "Guideline on computerised systems and electronic data in clinical trials" verabschiedet, die im September 2023 in Kraft trat.
Der Arbeitskreis Medizinischer Ethik-Kommissionen (AKEK) hat eine aktuelle Handreichung zum Ehegattennotvertretungsrecht herausgegeben.