Research Group "Genetic basis of immunodeficiency"
Prof. Dr. Bodo GrimbacherThe research group „Genetic basis of immunodeficiency“ focuses on the genetics and molecular pathophysiology of primary immunodeficiencies.
Research Areas
The Molecular and Genetic Causes of Primary Immunodeficiencies
Many primary immunodeficiencies represent an “Experiment of Nature” (Robert Good), by having genetic alterations in their genomic code impairing immunocompetence. Interestingly enough, in recent years it became clear that:
- The very same mutation causing the immunodeficiency can also cause immune dysregulation/autoimmunity/autoinflammation
- Mutations in one and the same gene can lead to many different phenotypes, and may often even have a substantially reduced penetrance
- A very similar phenotype in different individuals (even within the same family) may have different (mono-)genetic causes
Identification of monogenetic defects in patients with immunodeficiency and/or immune dysregulation
During my postdoctoral time at the NIH in Bethesda, Maryland, I learned under the guidance of my mentor Jennifer Puck how to positionally clone candidate genes for Mendelian human diseases. Coming back to Freiburg as a young physician-scientist, I used my knowledge to identify the first monogenic cause (ICOS deficiency; Nat Immunol, 2003) for a condition known as CVID, which is the commonest symptomatic primary immunodeficiency in men. To date, CVID is known to be caused by at least 70 different monogenic causes, of which I contributed about 12 of them. I also identified the first monogenic cause for inflammatory bowel disease (IL10- and IL10-receptor deficiency; NEJM, 2009), and the first cause of monogenic susceptibility to fungal disease (CARD9 deficiency; NEJM, 2009). I also helped to identify the genetic cause for Kostmann’s disease (HAX1 deficiency; Nat Genet, 2007). More recently, I discovered that monogenic mutation in CTLA4 (Nat. Med. 2014) lead to a multi-organ immune dysregulation including immunodeficiency, and that mutations in NFKB1 (AJHG, 2015) or NFKB2 (Front Immunol, 2019) may also lead to severe immune dysregulation. In this tradition, we continue to contribute to the ever-growing list of monogenic causes for inborn errors of immunity, which currently (2024) encompasses almost 500 disease genes.
References
- Rolles B, et al. Telomere biology disorders may manifest as common variable immunodeficiency (CVID). Clin Immunol. 2023;257:109837. doi: 10.1016/j.clim.2023.109837.
- Peng XP, et al. Next generation sequencing (NGS)-based approach to diagnosing Algerian patients with suspected inborn errors of immunity (IEIs). Clin Immunol. 2023;256:109758. doi: 10.1016/j.clim.2023.109758.
- Fliegauf M, et al. Detrimental NFKB1 missense variants affecting the Rel-homology domain of p105/p50. Front Immunol. 2022;13:965326. doi: 10.3389/fimmu.2022.965326.
- Rojas-Restrepo J, et al. Establishing the Molecular Diagnoses in a Cohort of 291 Patients With Predominantly Antibody Deficiency by Targeted Next-Generation Sequencing: Experience From a Monocentric Study. Front Immunol. 2021;12:786516. doi: 10.3389/fimmu.2021.786516.
- Frede N, et al. Genetic Analysis of a Cohort of 275 Patients with Hyper-IgE Syndromes and/or Chronic Mucocutaneous Candidiasis. J Clin Immunol. 2021;41(8):1804-38. doi: 10.1007/s10875-021-01086-4.
- Fliegauf M, et al. A Pathogenic Missense Variant in NFKB1 Causes Common Variable Immunodeficiency Due to Detrimental Protein Damage. Front Immunol. 2021;12:621503. doi: 10.3389/fimmu.2021.621503.
- Eskandarian Z, et al. Assessing the Functional Relevance of Variants in the IKAROS Family Zinc Finger Protein 1 (IKZF1) in a Cohort of Patients With Primary Immunodeficiency. Front Immunol. 2019;10:568. doi: 10.3389/fimmu.2019.00568.
- Klemann C, et al. Clinical and Immunological Phenotype of Patients With Primary Immunodeficiency Due to Damaging Mutations in NFKB2. Front Immunol. 2019;10:297. doi: 10.3389/fimmu.2019.00297.
- Frey-Jakobs S, et al. ZNF341 controls STAT3 expression and thereby immunocompetence. Sci Immunol. 2018;3(24) doi: 10.1126/sciimmunol.aat4941.
- Petersen BS, et al. Targeted Gene Panel Sequencing for Early-onset Inflammatory Bowel Disease and Chronic Diarrhea. Inflamm Bowel Dis. 2017;23(12):2109-20. doi: 10.1097/mib.0000000000001235.
- Schubert D, et al. Plasma cell deficiency in human subjects with heterozygous mutations in Sec61 translocon alpha 1 subunit (SEC61A1). J Allergy Clin Immunol. 2018;141(4):1427-38. doi: 10.1016/j.jaci.2017.06.042.
- Schepp J, et al. Screening of 181 Patients With Antibody Deficiency for Deficiency of Adenosine Deaminase 2 Sheds New Light on the Disease in Adulthood. Arthritis Rheumatol. 2017;69(8):1689-700. doi: 10.1002/art.40147.
- Volk T, et al. DCLRE1C (ARTEMIS) mutations causing phenotypes ranging from atypical severe combined immunodeficiency to mere antibody deficiency. Hum Mol Genet. 2015;24(25):7361-72. doi: 10.1093/hmg/ddv437.
- Elgizouli M, et al. Activating PI3Kδ mutations in a cohort of 669 patients with primary immunodeficiency. Clin Exp Immunol. 2016;183(2):221-9. doi: 10.1111/cei.12706.
- Fliegauf M, et al. Haploinsufficiency of the NF-κB1 Subunit p50 in Common Variable Immunodeficiency. Am J Hum Genet. 2015;97(3):389-403. doi: 10.1016/j.ajhg.2015.07.008.
- Dziadzio M, et al. Symptomatic males and female carriers in a large Caucasian kindred with XIAP deficiency. J Clin Immunol. 2015;35(5):439-44. doi: 10.1007/s10875-015-0166-0.
- Schubert D, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med. 2014;20(12):1410-6. doi: 10.1038/nm.3746.
- Sassi A, et al. Hypomorphic homozygous mutations in phosphoglucomutase 3 (PGM3) impair immunity and increase serum IgE levels. J Allergy Clin Immunol. 2014;133(5):1410-9, 9.e1-13. doi: 10.1016/j.jaci.2014.02.025.
- Lanternier F, et al. Deep dermatophytosis and inherited CARD9 deficiency. N Engl J Med. 2013;369(18):1704-14. doi: 10.1056/NEJMoa1208487.
- Lopez-Herrera G, et al. Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet. 2012;90(6):986-1001. doi: 10.1016/j.ajhg.2012.04.015.
- Glocker EO, et al. Infant colitis--it's in the genes. Lancet. 2010;376(9748):1272. doi: 10.1016/s0140-6736(10)61008-2.
- Woellner C, et al. Mutations in STAT3 and diagnostic guidelines for hyper-IgE syndrome. J Allergy Clin Immunol. 2010;125(2):424-32.e8. doi: 10.1016/j.jaci.2009.10.059.
- Engelhardt KR, et al. Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal-recessive form of hyper-IgE syndrome. J Allergy Clin Immunol. 2009;124(6):1289-302.e4. doi: 10.1016/j.jaci.2009.10.038.
- Glocker EO, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. 2009;361(21):2033-45. doi: 10.1056/NEJMoa0907206.
- Glocker EO, et al. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N Engl J Med. 2009;361(18):1727-35. doi: 10.1056/NEJMoa0810719.
- Salzer U, et al. Relevance of biallelic versus monoallelic TNFRSF13B mutations in distinguishing disease-causing from risk-increasing TNFRSF13B variants in antibody deficiency syndromes. Blood. 2009;113(9):1967-76. doi: 10.1182/blood-2008-02-141937.
- Salzer U, et al. Sequence analysis of BIRC4/XIAP in male patients with common variable immunodeficiency. Int Arch Allergy Immunol. 2008;147(2):147-51. doi: 10.1159/000135702.
- Salzer U, et al. Screening of functional and positional candidate genes in families with common variable immunodeficiency. BMC Immunol. 2008;9:3. doi: 10.1186/1471-2172-9-3.
- Pfeifer D, et al. The hyper-IgE syndrome is not caused by a microdeletion syndrome. Immunogenetics. 2007;59(12):913-26. doi: 10.1007/s00251-007-0257-z.
- Holland SM, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med. 2007;357(16):1608-19. doi: 10.1056/NEJMoa073687.
- Salzer U, et al. Sequence analysis of TNFRSF13b, encoding TACI, in patients with systemic lupus erythematosus. J Clin Immunol. 2007;27(4):372-7. doi: 10.1007/s10875-007-9094-y.
- Klein C, et al. HAX1 deficiency causes autosomal recessive severe congenital neutropenia (Kostmann disease). Nat Genet. 2007;39(1):86-92. doi: 10.1038/ng1940.
- Bohn G, et al. A novel human primary immunodeficiency syndrome caused by deficiency of the endosomal adaptor protein p14. Nat Med. 2007;13(1):38-45. doi: 10.1038/nm1528.
- Finck A, et al. Linkage of autosomal-dominant common variable immunodeficiency to chromosome 4q. Eur J Hum Genet. 2006;14(7):867-75. doi: 10.1038/sj.ejhg.5201634.
- Jung J, et al. Identification of a homozygous deletion in the AP3B1 gene causing Hermansky-Pudlak syndrome, type 2. Blood. 2006;108(1):362-9. doi: 10.1182/blood-2005-11-4377.
- Schäffer AA, et al. Analysis of families with common variable immunodeficiency (CVID) and IgA deficiency suggests linkage of CVID to chromosome 16q. Hum Genet. 2006;118(6):725-9. doi: 10.1007/s00439-005-0101-1.
- Salzer U, et al. Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nat Genet. 2005;37(8):820-8. doi: 10.1038/ng1600.
- Salzer U, et al. ICOS deficiency in patients with common variable immunodeficiency. Clin Immunol. 2004;113(3):234-40. doi: 10.1016/j.clim.2004.07.002.
- Grimbacher B, et al. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat Immunol. 2003;4(3):261-8. doi: 10.1038/ni902.
Funding
This project is funded by internal funds of the Medical Center – University of Freiburg.
Studying DNA histone modifications as determinant for infection susceptibility
Cleavage under targets and release using nuclease (CUT & RUN)
To comprehensively understand the contributions of histone modifications to transcript regulation, we generated data on three CVID patients (5000026, 5001414, and 5000291) and three controls (FR124, FR135, and FR145). We performed Cleavage under targets and release using nuclease (CUT & RUN) to analyze histone modifications of active (H3K27ac) and repressive chromatin (H3K27me3) derived from stimulated naïve B cells.
Materials and methods
Sample preparation. Peripheral Blood Mononuclear Cells (PBMCs) were isolated from EDTA blood by Ficoll density centrifugation following standard protocols. A total of three patient and three control PBMC samples were sorted into naïve B cells (CD19+, IgD, CD27-) and stimulated for 24 hours. Three patient and three control PBMC samples were sorted into naïve B cells (CD19+, IgD-, CD27-) and stimulated for 24 hours. Cells were sorted on a MoFlo Astrios Cell sorter (Beckman Coulter, Brea, CA, USA).
Cell stimulation. Naïve B lymphocytes were stimulated with 1µg/ml CD40L (provided by Pascal Schneider, Lausanne, Switzerland) and 50ng/ml IL21 and incubated for 24 hours at 37ºC, 5 % CO2. After stimulation, cell numbers were determined with a Neubauer chamber. A total of 5,000-50,000 cells were used for CUT&RUN processing. B cell activation (CD69 APC, CD80 Brilliant Violet 421, CD86 PerCP-Cy5.5, CD95 Brilliant Violet 650, HLA-DR PE-Cy7) was measured on a LSR Fortessa from Becton Dickinson (BD Biosciences, San Jose, CA).
CUT&RUN protocol. Samples were processed according to a modified protocol and available on protocols.io (1). Antibodies used included H3K27ac (Abcam, 4729), H3K27me3 (Cell Signalling, 9733), and IgG (Cell Signaling, 2729). Samples from three CVID patients and three controls were used for each antibody. CUT&RUN library preparation and sequencing was performed by Novogene (Cambridge, UK), using the HiSeq (Illumina) in paired-end mode with reads of 150bp length.
Bioinformatics analysis. Detailed bioinformatics analysis workflows with command line examples for illumina sequences are available on protocols io (2). Data preprocessing was performed on the Galaxy web platform and the public server usegalaxy.eu. Briefly, trimmed fastq files were aligned to the hg38 reference genome using Bowtie 2. Aligned reads were extracted and converted to a sorted bam file using SAM tools. Scaled bigwig files were generated using Deeptools, and peaks were called using Macs2. Further analysis was performed in R v4.2.0 using a custom R script. For annotation, BED files of the identified peaks were loaded and harmonized using the GenomicRanges and rtracklayer packages in R. Default chromosome names (chr1-chr22, chrX, chrY) were used. Feature counts for the identified peaks were generated using the featureCounts function from the Rsubread package. Count matrices were generated and normalized for subsequent analysis. Differential binding analysis was performed using the DESeq2 package in R. Count data were normalized and differential peaks between CVID patients and controls were identified. Genomic annotation bar plots and pie charts were generated using the plotAnnoBar and plotAnnoPie functions of the ChIPseeker package. These plots showed the distribution of peaks across genomic features such as promoters, exons, introns, 5′ UTR, 3′ UTR, downstream regions, and intergenic regions. Significantly differentially bound regions were identified and annotated. Their potential regulatory roles were inferred based on their genomic locations and annotations.
Results
We investigated the differential binding patterns of two key histone modifications, H3K27ac and H3K27me3, in CVID patients compared to controls. H3K27ac is associated with active enhancers and promoters, indicating regions of active transcription, while H3K27me3 is associated with gene repression, indicating silenced regions of the genome. A total of 18 samples were sent for sequencing One control sample failed library sequencing (FR135), resulting in the analysis of fifteen stimulated naïve B cell samples.
H3K27ac binding patterns
The comparative analysis of the genomic distribution of H3K27ac binding sites between healthy controls and CVID patients showed significant differences (Figure 1a). In healthy controls, the majority of H3K27ac binding sites are located in distal intergenic regions (85.73 %), suggesting a dominant role for distal enhancers in regulating gene expression. In contrast, CVID patients show a reduced proportion of binding sites in distal intergenic regions (50.82 %) and an increased proportion in promoter regions (<=3kb) and introns. Specifically, CVID patients have more binding sites in promoter regions (3.86 % for <=1kb, 3.42 % for 1-2kb, and 4.46 % for 2-3kb) and in introns (11.89 % for 1st intron and 22.29 % for other introns). These differences suggest a shift in the regulatory landscape in CVID patients. On the differential analysis, we found 27 significant differential binding regions (Figure 1b). Specifically, NUP88, involved in nucleocytoplasmic transport, and DUSP8, a regulator of MAPK signaling pathways, showed reduced binding (log2 fold change ~ -4 and -3.5, respectively). On the other hand, BMI1, part of the Polycomb Repressive Complex 1 (PRC1) involved in gene silencing showed decreased H3K27ac binding in CVID patients (log2 fold change ~ -2.7). These changes suggest a significant impact on enhancer and promoter activity, which may lead to dysregulated gene expression and impaired immune responses in CVID patients.
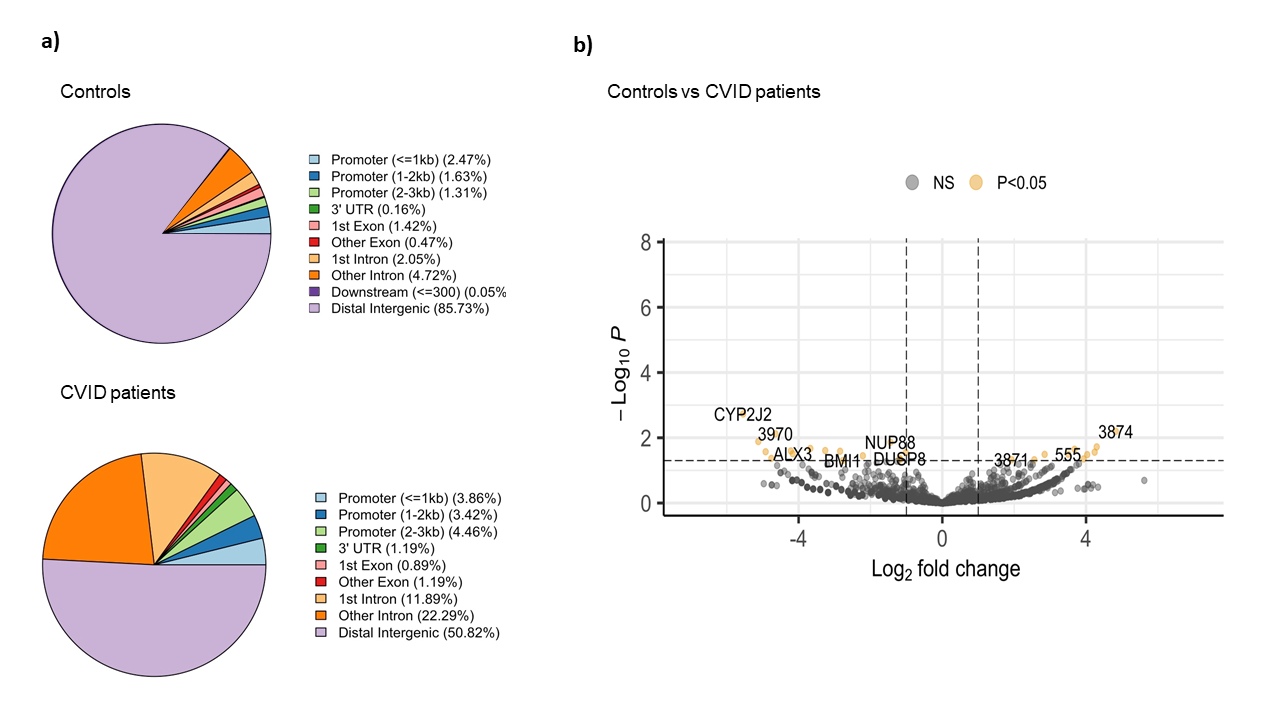
Figure 1: Binding analysis of H3K27ac on stimulated naïve B cells between CVID patients and controls. a) Pie charts show the distribution of H3K27ac binding sites across different genomic regions in CVID patients and healthy controls. b) The volcano plot shows the differential binding of H3K27ac. The x-axis represents the log2 fold change in binding intensity between CVID patients and healthy controls, while the y-axis indicates the -log10 (p-value) of the differences. Points are color coded according to their significance: non-significant (NS) in grey, significant log2 fold change with p-value (P < 0.05) in yellow. Genes with significant differential binding are labelled, as well as regions where no specific gene name was found (shown as number).
H3K27me3 binding patterns
The comparative analysis of the genomic distribution of H3K27me3 binding sites between controls and CVID patients revealed significant differences (Figure 2a). In healthy controls, a significant proportion of H3K27me3 binding sites are located in distal intergenic regions (71.04 %), suggesting a predominant role in long-range chromatin interactions that mediate gene silencing. On the other hand, in CVID patients, the proportion of H3K27me3 binding sites in distal intergenic regions is reduced to 54.42 %. This shift is accompanied by an increased presence of H3K27me3 binding sites in promoter regions (3.45 % for <=1kb) and intronic regions (10.77 % for 1st intron and 21.69 % for other introns). These changes suggest a redistribution of repressive marks, possibly leading to altered gene silencing and transcriptional regulation in CVID patients. In CVID, there may be a disruption in the maintenance of a repressive chromatin state. We also found significant differential binding between CVID patients and healthy controls across 112 genomic regions (Figure 2b). Key affected genes include OCRL, involved in inositol phosphate metabolism, which showed a significant decrease in H3K27me3 binding (log2 fold change ~ -5). EEF1D, a translation elongation factor essential for protein synthesis, also showed reduced binding (log2 fold change ~ -4.5). In contrast, RNR1, essential for ribonucleotide reduction and DNA synthesis, showed increased H3K27me3 binding in CVID patients (log2 fold change ~ 5). These findings suggest altered gene silencing mechanisms, potentially leading to inappropriate gene regulation.
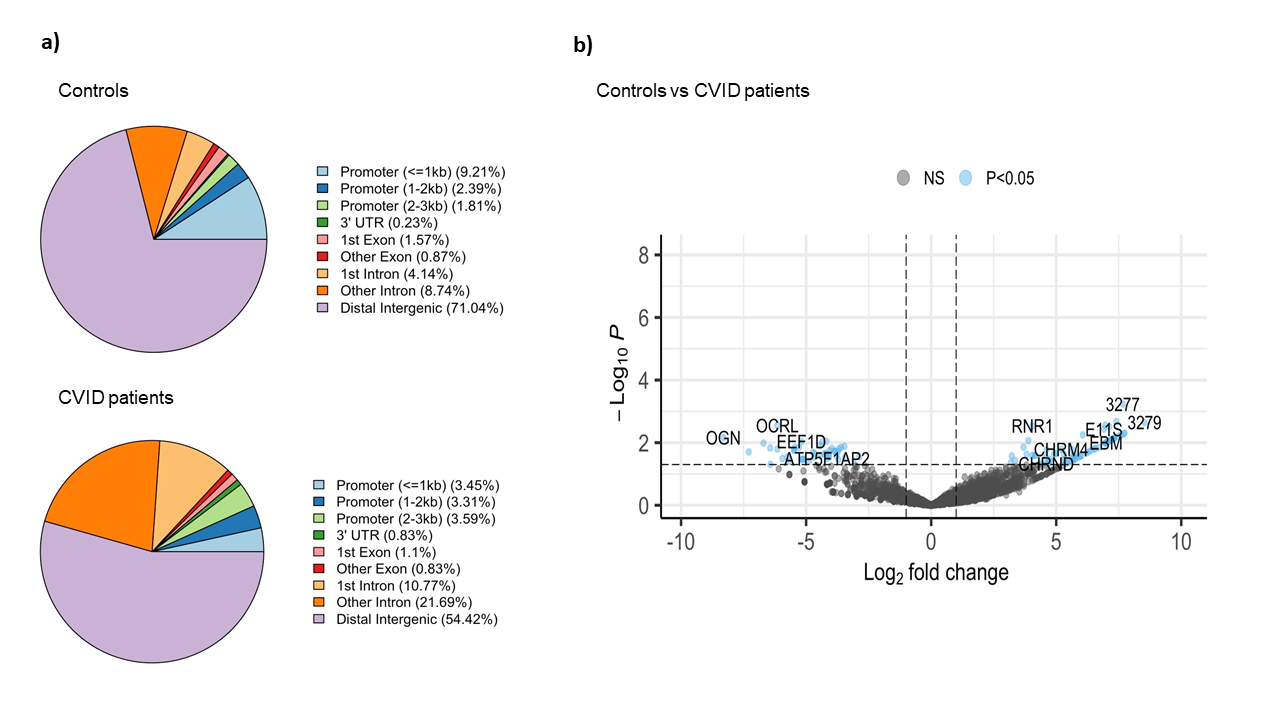
Figure 2: Binding analysis of H3K27me3 binding sites on stimulated naïve B cells in CVID patients and healthy controls. a) Pie chart show the distribution of H3K27me3 binding sites across different genomic features in controls and CVID patients. b) The volcano plot illustrates the differential binding of H3K27me3. The x-axis represents the log2 fold change in binding intensity between CVID patients and controls, while the y-axis indicates the -log10 (p-value) of the differences. Points are color coded according to their significance: non-significant (NS) in grey and significant log2 fold change with p-value (P < 0.05) in blue. Genes with significant changes are indicated.
In summary, the shift of H3K27ac binding from distal enhancers to promoter regions and introns in CVID patients indicates impaired enhancer and promoter activity leading to dysregulated gene expression. Similarly, the redistribution of H3K27me3 binding suggests impaired gene repression mechanisms, contributing to inappropriate gene regulation.
References
- Skene PJ, et al. Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat Protoc. 2018;13(5):1006-19. doi: 10.1038/nprot.2018.015.
- Schmitt A, et al. CUT&RUN chromatin profiling of human kidney tissue. protocols.io. www.protocols.io/view/cut-amp-run-chromatin-profiling-of-human-kidney-ti-bp2l615o1vqe/v1.Published 2022. Accessed May 6, 2024.
Funding
This project is primarily funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy – EXC 2155 “RESIST” – Project ID 390874280.
The role of the gut microbiota for the mammalian immune system – using inborn errors of immunity as a window into co-development and complexity
Inborn errors of immunity (IEI) comprise a group of more than 500 genetic disorders that result in the impairment of host immune responses. IEI display a broad range of manifestations, including recurrent infections, autoimmunity, lymphoproliferation, malignancy, and granulomas, posing challenges for physicians in diagnosis and treatment. Furthermore, the penetrance, expressivity, and severity of IEI vary even among patients carrying the same mutation. However, modifiers determining the disease penetrance, severity, and expressivity remain unknown.
The microbiome appears essential for maintaining host immunity (1). Therefore, especially in patients with a defective immune system, this delicate balance between immune homeostasis and microbial diversity is at stake. Hence, the microbial composition and diversity patients with inborn errors of immunity are under close investigation. Dysbiosis and reduced alpha-diversity are observed particularly in common variable immunodeficiency (CVID), chronic granulomatous disease (CGD) (2-4). However, whether microbial dysbiosis is the key determinant of IEI disease expressivity and severity remains unknown. Answering this question will encourage the development of novel therapies, such as fecal microbiota transplantation (FMT) (5), to restore the balance of the gut microbiota and improve IEI patient fitness.
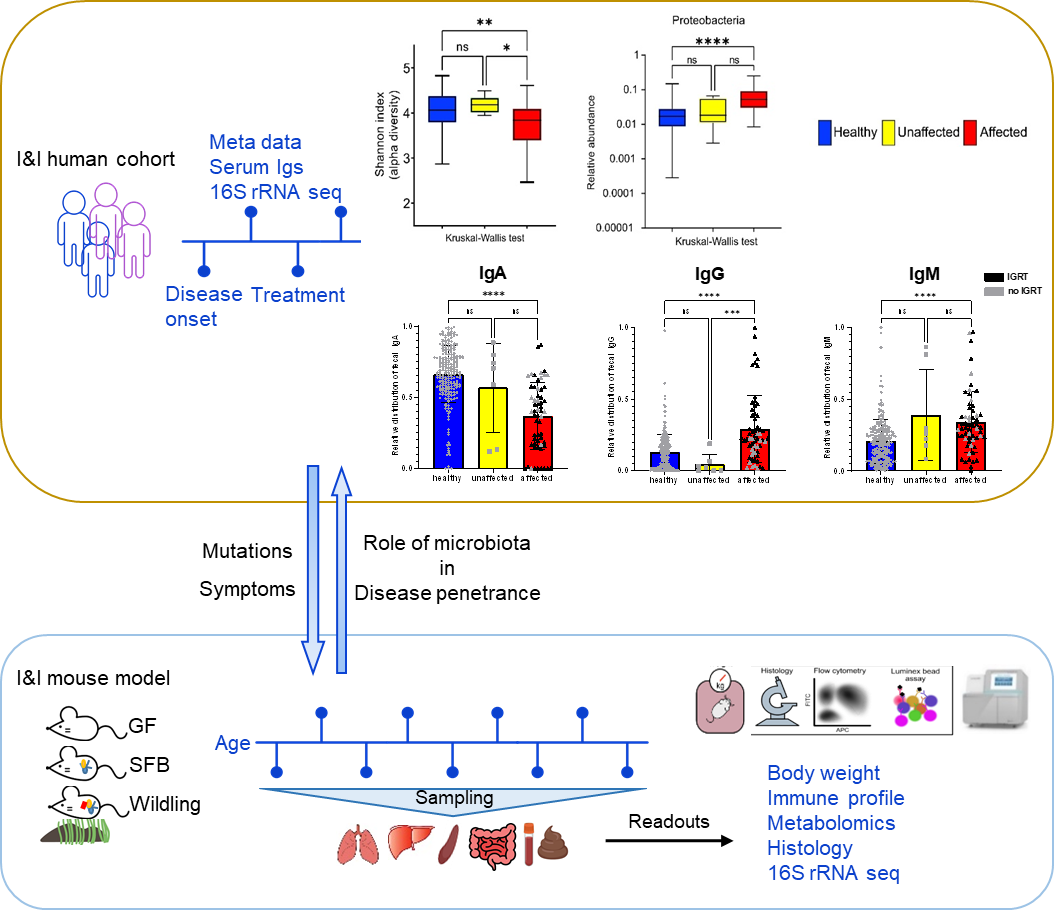
We combine human cohort studies and mouse models to investigate the role of the microbiota in disease penetrance and severity in patients with inborn errors of immunity, i.e. CTLA-4 insufficiency or NF-κB insufficiency. Through shotgun metagenomics and 16S rRNA sequencing of patient stool samples at different disease stages, we unveil how microbial dysbiosis develops with disease progression. Moreover, by evaluating the immune landscape in ctla4+/- or nfkb+/- gnotobiotic and wildling mice, we seek to elucidate the immune pathophysiology of CTLA4 insufficiency or NF-κB insufficiency and determine whether the microbial dysbiosis is cause or consequence of the immune dysregulation.
References
- Zhao B, et al. Helicobacter spp. are prevalent in wild mice and protect from lethal Citrobacter rodentium infection in the absence of adaptive immunity. Cell Rep. 2023;42(6):112549. doi: 10.1016/j.celrep.2023.112549.
- Sharma M, et al. Microbiome and Its Dysbiosis in Inborn Errors of Immunity. Pathogens. 2023;12(4) doi: 10.3390/pathogens12040518.
- Nöltner C, et al. Fecal Immunoglobulin Levels as a Modifier of the Gut Microbiome in Patients with Common Variable Immunodeficiency. J Clin Immunol. 2023;43(6):1208-20. doi: 10.1007/s10875-023-01469-9.
- van Schewick CM, et al. Altered Microbiota, Impaired Quality of Life, Malabsorption, Infection, and Inflammation in CVID Patients With Diarrhoea. Front Immunol. 2020;11:1654. doi: 10.3389/fimmu.2020.01654.
- Egg D, et al. Therapeutic options for CTLA-4 insufficiency. J Allergy Clin Immunol. 2022;149(2):736-46. doi: 10.1016/j.jaci.2021.04.039.
Funding
This study is supported by the German Research Foundation (DFG), collaborative research center IMPATH, project SFB1160/3_B5.
Molecular and cellular effects of pathogenic NFKB1 and NFKB2 sequence variants in primary antibody deficiency syndromes
Common variable immunodeficiency (CVID) is the most prevalent symptomatic primary antibody deficiency (PAD), with recurrent infections, mostly in the upper respiratory tract. Genetic defects are identified in approximately 20 % of CVID patients. Among these, heterozygous pathogenic variants in NFKB1 or NFKB2 collectively account for up to 10 % of the monogenic forms. NFKB1 encodes the NF-kappaB1 (NF-κB1) transcription factor precursor p105 which is processed to p50 (canonical pathway), whereas NFKB2 encodes p100/p52 (non-canonical pathway). The dimeric transcription factors of the NF-κB family are composed of a variety of hetero- and homodimers of the subunits p50, p52 and the three Rel-proteins RelA (=p65), RelB and cRel, play important roles in numerous biological processes including the immune system and mediate transcriptional responses to a multitude of stimuli.
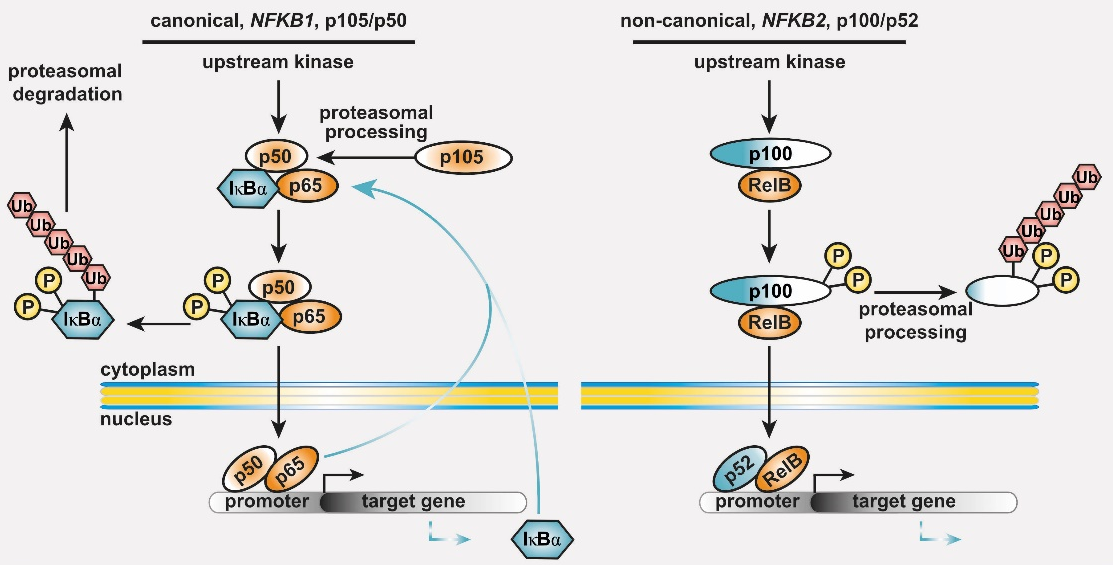
Figure 1: The basic principles of the NF-κB signaling systems. The NF-κB signaling network is highly complex and operative in most cell types. (left) The canonical NF-κB1 pathway integrates signals from receptors (TCR, BCR, TNF-R and TLRs in lymphocytes), co-stimulators, and metabolites (e.g. glutamine). Processing of its C-terminal half converts the NF-κB1 precursor p105 into the transcription factor subunit p50, which assembles with RelA (also known as p65) and the inhibitory protein IκBα an inactive cytoplasmic trimer. Pathway stimulation activates upstream kinases, which leads to phosphorylation, polyubiquitination and degradation of IκBα. Subsequently, the preassembled transcription factor p50/RelA is released, translocates to the nucleus, and regulates its target genes. (right) Activation of the non-canonical NF-κB2 pathway occurs through BAFF receptor signaling, CD40 or the lymphotoxin-beta receptor. Pathway stimulation leads to proteasomal degradation of the inhibitory C-terminal half of the p100 precursor thereby releasing the active p52/RelB transcription factor. Homodimers of p50/p50 and p52/p52 are not equipped with a transactivation domain and act as transcriptional repressors (not shown). Both cytoplasmic precursor proteins administrate IκB-like activity on the transcription factor dimers.
In vitro analysis of protein defects associated with NFKB1 and NFKB2 sequence variants
We are using a cell-culture-based transient overexpression model to evaluate the pathogenic relevance of NFKB1 and NFKB2 sequence variants. Following site-directed mutagenesis, the ectopically expressed mutant proteins are analyzed for functional defects. The testing system solely relies on the knowledge of a given variant e.g. from genetic counseling, blood sampling is not required. According to the ‘type’ of the identified protein defect, we assign the variants into distinct categories.
The most-common ‘type’ of the known pathogenic NFKB1 variants affects the N-terminal half of the protein (typically on the N-terminal side relative to the nuclear localization sequence, NLS) and predicts the expression of severely truncated, non-functional proteins (if expressed at all) which undergo intensified decay and cause (haplo-)insufficiency of both, p105 and p50. Truncating variants in the central/proximal part of the protein predict skipping of the precursor stage and immediate expression of p50-like proteins. Rarely, truncating variants affect the C-terminal domains of p105, yet with unknown effects. Internal deletions result from splicing errors. A variety of known pathogenic missense variants (single amino acid changes), all residing within the N-terminal Rel-homology domain (RHD), cause protein-decay or, less frequent, loss-of-DNA-binding activity. The relevance of the frequently occurring missense variants within the C-terminal half of p105 (the part which is removed upon generation of p50) is largely unknown.
Most of the known disease-causing NFKB2 variants cause expression of non-processable p100 precursor proteins – and thus an excess of the associated IκB-like activity – and, as a ‘side-effect’, insufficiency of p52. Also known are (i) proximally truncating precursor-skipping variants predicting the expression of p52-like nuclear proteins, (ii) short truncations associated with haploinsufficiency of p100 and p52, and (iii) diverse missense variants with yet unpublished effects.
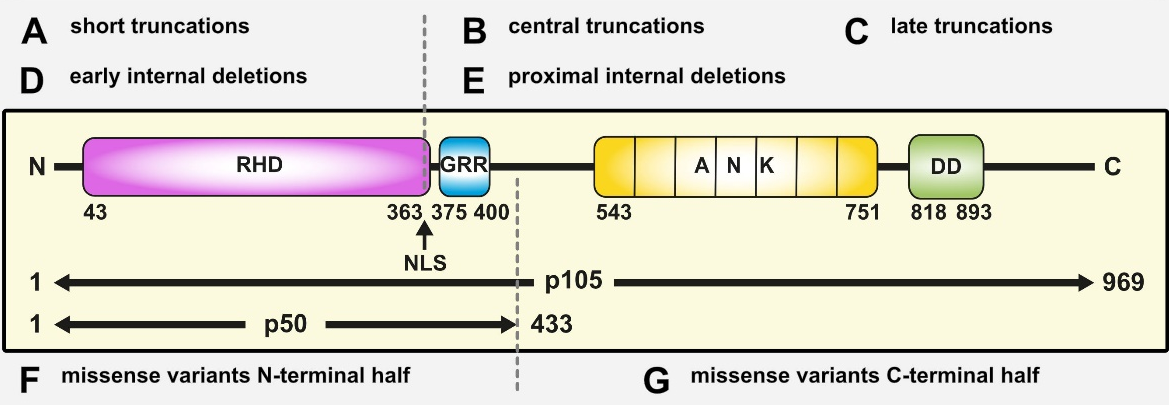
Figure 2: Pathogenic NFKB1 variants cause diverse protein defects. Schematic protein structure of the p105 precursor with the N-terminal Rel homology domain (RHD), the central nuclear localization sequence (NLS) and the glycine-rich region (GRR), and the C-terminal Ankyrin-repeat domain (ANK) and the death domain (DD). The mature p50 comprises the N-terminal half of p105. Numbers indicate amino acid positions. Depending on the ‘type’ of protein defect, miscellaneous signaling processes might be deranged. Due to the complexity of NF-kB signaling, this concept is not exhaustive (adapted from Fliegauf & Grimbacher JACI 2018;142(4):1062-1065.)
NFKB1 and NFKB2-omics
The transcription factor NF-κB plays a pivotal role in the adaptive immune response. Pathogenic variants in NFKB1 are the most common genetic etiology of common variable immunodeficiency (CVID). Patients frequently present with impaired terminal B cell differentiation, autoimmunity, and hyperinflammatory immune dysregulation. NF-κB signaling and target gene expression are expected to be dysregulated in NFKB1-mutated patients. Here, we performed a multi-omics characterization of lymphocytes and monocytes from a cohort of clinically affected and unaffected NFKB1 mutation carriers. Our analysis of B lymphocytes identified specific epigenetic dysregulation and gene expression differences on B cells from NFKB1-mutated patients. We observed an aberrant expression of negative regulators of NF-κB signaling in NFKB1 mutation carriers, which may be a key factor for the autoinflammatory phenotype of these patients. Moreover, our analysis points towards a dysregulation of XBP1 and BCL3, key players of B cell activation and proliferation at different stages of B cell differentiation. The analysis of T cells and monocytes is still pending.
The reduced expression of negative regulators of the NF-κB network is likely to be one of several mechanisms responsible for the aberrant NF-κB signaling, which impairs the maintenance of a normal humoral immune response.
In summary, our findings highlight epigenetic and gene expression changes in B cells associated with NFKB1 mutations. Our data give insight into future therapeutic opportunities for patients with NFKB1 (haplo)insufficiency.
Phenotypic characterization, disease history, and treatment outcome of patients carrying pathogenic variants in NFKB1 or NFKB2
Analysis of the clinical and immunological phenotype of affected patients with pathogenic heterozygous NFKB1 variants has revealed an incomplete penetrance and an age-dependent disease severity. Patients are characterized by hypo-gammaglobulinemia, reduced switched memory B cells and recurrent respiratory and gastrointestinal infections. In addition, autoimmunity, lymphoproliferation, non-infectious enteropathy, opportunistic infections, autoinflammation and malignancy are frequently observed. Increased susceptibility to bacterial, viral and fungal infections is typical and autoantibodies are detectable in a rather large proportion.
Despite an unmet medical need, no targeted treatment is available for this rare condition. Current treatment protocols include immunoglobulin replacement and antibiotic treatment, as well as application of immunosuppressive or anti-inflammatory agents such as anti-IL1 and anti-TNF-α. Moreover, abatacept (a CTLA4 fusion protein) appears to be a promising option.
We therefore investigate, whether the observed autoinflammation and the increased frequency of viral infections (both reminiscent of interferonopathies) is associated with an altered type 1 interferon expression pattern in innate immune cells. Since the observed autoimmune disease is indicative of defective regulatory T cells (Tregs), we also aim at a detailed characterization and phenotyping of Tregs (and effector T cell subsets) in patients with pathogenic NFKB1 variants. In addition, we are testing whether the increased frequency of cytopenia in these patients is mediated by autoantibodies. We furthermore analyze whether a second genetic alteration (either somatic or germline) accounts for the observed incomplete and age-dependent penetrance.
To compare the effect of different therapeutic options, we have developed an NFKB1 Disease Activity Score (NFKB1-DAS). Using the score, the therapeutic success can be measured objectively in clinical trials. The score can also be used for clinical management to monitor the course of disease activity in patients. Based on data from the literature and data from patients in Germany and Switzerland, we decided which disease manifestations the score should reflect. The final score consists of 21 parameters, which depict the various organ manifestations of the disease, according to their severity. Further validation of the score is currently performed based on a global cohort of patients with NFKB1 insufficiency and its feasibility is being tested in practice. Treatment options such as anti-TNF-α, anti-IL1, or JAK-inhibition are currently being analysed using the NFKB1-DAS.
From many years of caring for our patients, we know that NFKB1 insufficiency may lead to a relevant impairment of quality of life. In our clinical research, we therefore implement questionnaires on quality of life and use these to evaluate the various treatment options, in addition to the NFKB1-DAS.
If you are interested in cooperating with us on our projects, for example by contributing patient data, or if you are interested in the NFKB1 Disease Activity Score, please contact Prof. Dr. Bodo Grimbacher.
The contributions of our group to the field of NF-κB research are the following:
- Le Voyer T, et al. Autoantibodies against type I IFNs in humans with alternative NF-κB pathway deficiency. Nature. 2023;623(7988):803-13. doi: 10.1038/s41586-023-06717-x.
- Camacho-Ordonez N, et al. Integrated Multi-omics Analyses of NFKB1 patients B cells points towards an up regulation of NF-κB network inhibitors. bioRxiv. 2022:2022.11.22.517350. doi: 10.1101/2022.11.22.517350.
- Fliegauf M, et al. Detrimental NFKB1 missense variants affecting the Rel-homology domain of p105/p50. Front Immunol. 2022;13:965326. doi: 10.3389/fimmu.2022.965326.
- Rojas-Restrepo J, et al. Establishing the Molecular Diagnoses in a Cohort of 291 Patients With Predominantly Antibody Deficiency by Targeted Next-Generation Sequencing: Experience From a Monocentric Study. Front Immunol. 2021;12:786516. doi: 10.3389/fimmu.2021.786516.
- Keller B, et al. The expansion of human T-bet(high)CD21(low) B cells is T cell dependent. Sci Immunol. 2021;6(64):eabh0891. doi: 10.1126/sciimmunol.abh0891.
- Bergbreiter A, et al. Recurrent necrotizing cellulitis, multi-organ autoimmune disease and humoral immunodeficiency due to a novel NFKB1 frameshift mutation. Eur J Med Genet. 2021;64(3):104144. doi: 10.1016/j.ejmg.2021.104144.
- Li J, et al. Biochemically deleterious human NFKB1 variants underlie an autosomal dominant form of common variable immunodeficiency. Journal of Experimental Medicine. 2021;218(11) doi: 10.1084/jem.20210566.
- Fliegauf M, et al. A Pathogenic Missense Variant in NFKB1 Causes Common Variable Immunodeficiency Due to Detrimental Protein Damage. Front Immunol. 2021;12:621503. doi: 10.3389/fimmu.2021.621503.
- Lorenzini T, et al. Characterization of the clinical and immunologic phenotype and management of 157 individuals with 56 distinct heterozygous NFKB1 mutations. J Allergy Clin Immunol. 2020;146(4):901-11. doi: 10.1016/j.jaci.2019.11.051.
- Schröder C, et al. Late-Onset Antibody Deficiency Due to Monoallelic Alterations in NFKB1. Frontiers in Immunology. 2019;10 doi: 10.3389/fimmu.2019.02618.
- Klemann C, et al. Clinical and Immunological Phenotype of Patients With Primary Immunodeficiency Due to Damaging Mutations in NFKB2. Frontiers in Immunology. 2019;11 doi: 10.3389/fimmu.2019.00297.
- Fliegauf M, Grimbacher B. Nuclear factor κB mutations in human subjects: The devil is in the details. Journal of Allergy and Clinical Immunology. 2018;142(4):1062-5. doi: doi.org/10.1016/j.jaci.2018.06.050.
- Lougaris V, et al. NFKB1 regulates human NK cell maturation and effector functions. Clin Immunol. 2017;175:99-108. doi: 10.1016/j.clim.2016.11.012.
- Lougaris V, et al. Early and late B-cell developmental impairment in nuclear factor kappa B, subunit 1-mutated common variable immunodeficiency disease. J Allergy Clin Immunol. 2017;139(1):349-52.e1. doi: 10.1016/j.jaci.2016.05.045.
- Keller B, et al. Disturbed canonical nuclear factor of κ light chain signaling in B cells of patients with common variable immunodeficiency. J Allergy Clin Immunol. 2017;139(1):220-31.e8. doi: 10.1016/j.jaci.2016.04.043.
- Fliegauf M, et al. Haploinsufficiency of the NF-κB1 Subunit p50 in Common Variable Immunodeficiency. Am J Hum Genet. 2015;97(3):389-403. doi: 10.1016/j.ajhg.2015.07.008.
Funding
This project is primarily funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy – CIBSS – EXC-2189 – Project ID 390939984.
Molecular mechanisms of the hyper IgE syndrome
The autosomal dominant hyper IgE syndrome (AD-HIES) is a rare primary immunodeficiency, caused mainly by heterozygous missense mutations in the signal transducer and activator of transcription 3 (STAT3) gene (1). STAT3 is activated after cytokine-induced signaling, and therefore is relevant in many lymphocyte functions and development like the differentiation of the T helper (Th)17 cells (2).
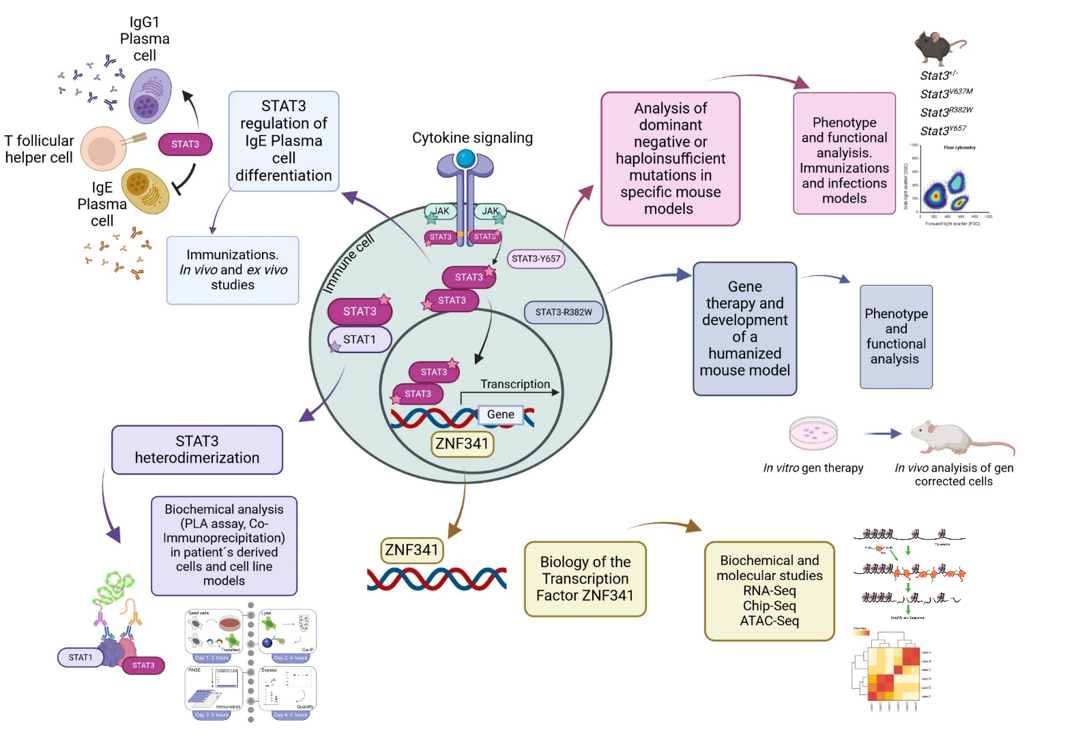
In our group, we investigate the mutations in STAT3 causing AD-HIES and how others transcription factors that interact or regulate STAT3 are involved in this process. By using a broad range of biochemical and molecular techniques, together with the development of mouse models, we evaluate the different mechanisms in which these mutations can affect the survival and differentiation of specific immune cells.
In particular, we are interested in evaluating different STAT3 mutations causing AD-HIES, and discovering new mechanisms that will explain the phenotype observed in patients, from having a dominant negative or haploinsufficient effect, to analyzing the heterodimer formation of STAT3 with other STAT molecule, and in evaluating how IgE+ plasma cells are generated in these patients. At the same time, we are studying how mutations in the transcription factor ZNF341, which produces a similar phenotype to AD-HIES (3), is regulating STAT3 and vice versa. Finally, we are developing humanized mouse models with patient’s derived cells that were modified by using gene therapy (4), to have a Wildtype STAT3 gene. This study in particular, and all in general, will give us new understanding of how STAT3 functions and will open new perspectives for the development of future therapies for AD-HIES patients.
References
- Holland SM, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med. 2007;357(16):1608-19. doi: 10.1056/NEJMoa073687.
- Ma CS, et al. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J Exp Med. 2008;205(7):1551-7. doi: 10.1084/jem.20080218.
- Frey-Jakobs S, et al. ZNF341 controls STAT3 expression and thereby immunocompetence. Sci Immunol. 2018;3(24) doi: 10.1126/sciimmunol.aat4941.
- König S, et al. Allele-Specific Disruption of a Common STAT3 Autosomal Dominant Allele Is Not Sufficient to Restore Downstream Signaling in Patient-Derived T Cells. Genes (Basel). 2022;13(10) doi: 10.3390/genes13101912.
Funding
This project is funded by the Deutsche Forschungsgemeinschaft (DFG) grant GR 1617/17-1, project number 519635399 and the EU-funded PhD program IMMERGE.
Investigating the benefit of selective PI3-kinase–δ (PIK3CD) inhibition for patients with immune dysregulation
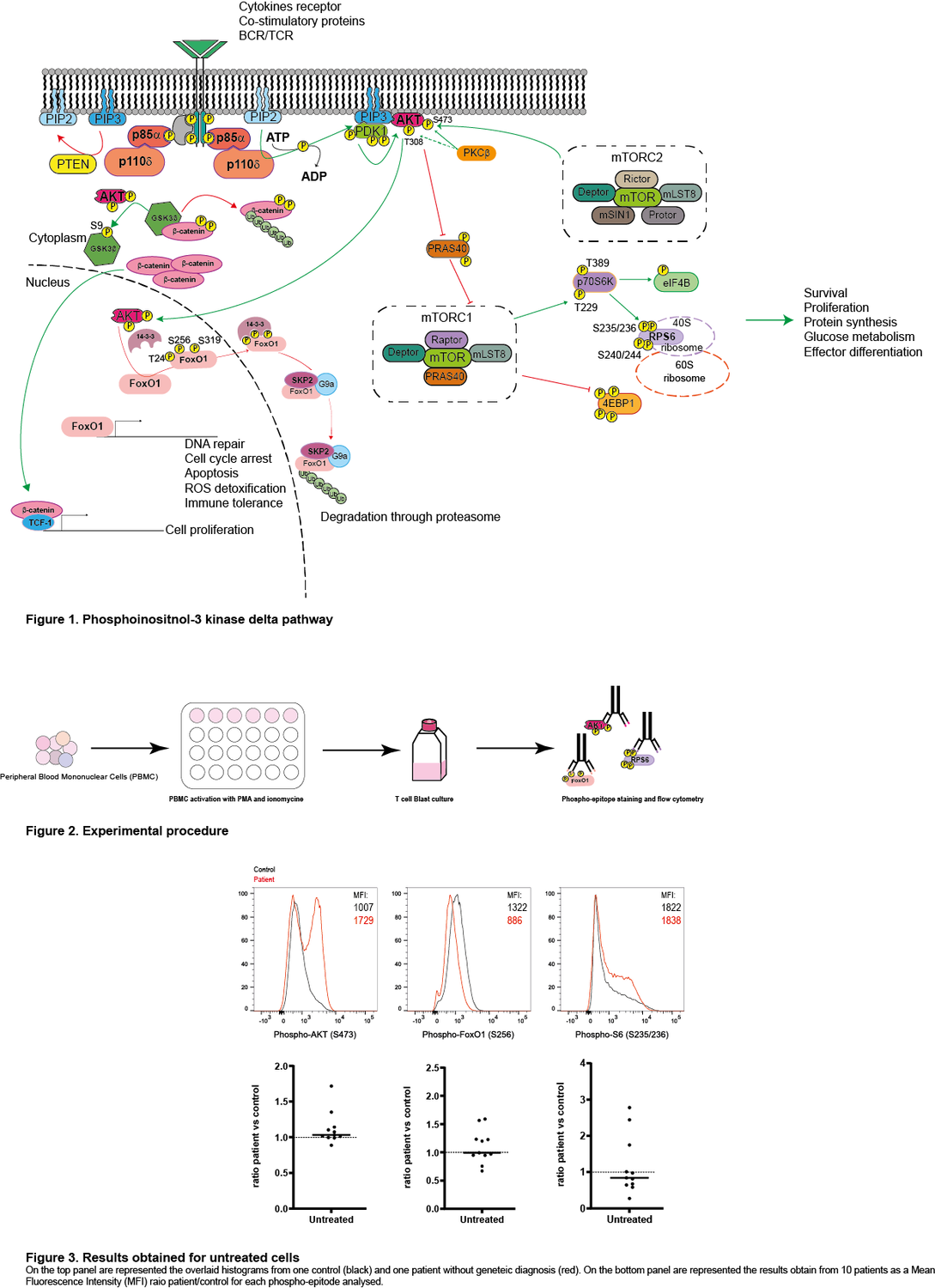
Class I phosphatidylinositol 3-kinase delta (PIK3δ) is composed of two subunits, the catalytic p110δ expressed only in lymphocytes and the ubiquitously expressed regulatory p85α. Activation of this pathway by the T-cell- or B-cell receptor (TCR/BCR), or cytokine receptors, leads to the production of phosphatidylinositol-3,4,5-trisphosphate (PIP3), phosphorylation of AKT, activation of mTORC1 and subsequent phosphorylation of the ribosomal S6 protein. This leads to protein synthesis and improved cell survival, proliferation and migration.
Humans may carry mutations in either of the two genes encoding the two subunits: PIK3CD encoding p110δ and PIK3R1 encoding p85α, leading to Activated p110δ Syndromes (APDS). Patients show a constitutive activation of the pathway, leading to an immune dysregulation syndrome. Pharmaceutical companies have succeeded to develop a PIK3δ-specific inhibitor (leniolisib) to treat patients with APDS.
Our lead question for this research project is: Could the specific PIK3δ inhibitor – leniolisib – be useful for other patients than APDS?
With financial support of the company Pharming, we are screening different patients with common variable immunodeficiency (CVID) with or without an identified mutation for the activation level of PIK3δ by analysing the phosphorylation of AKT, S6, and FOXO in T cell blasts. On the one hand, for the patients with a known mutation, we expect to see a specific pattern of PIK3δ activation and perhaps determine if these specific patients could benefit from leniolisib treatment. On the other hand, for the patients with an unknown cause of CVID, we expect to determine on a case-by-case basis whether the patient may benefit from PIK3δ inhibition.
References
- Deau MC, et al. A human immunodeficiency caused by mutations in the PIK3R1 gene. J Clin Invest. 2014;124(9):3923-8. doi: 10.1172/jci75746.
- Maccari ME, et al. Activated phosphoinositide 3-kinase δ syndrome: Update from the ESID Registry and comparison with other autoimmune-lymphoproliferative inborn errors of immunity. J Allergy Clin Immunol. 2023;152(4):984-96.e10. doi: 10.1016/j.jaci.2023.06.015.
- Maccari ME, et al. Disease Evolution and Response to Rapamycin in Activated Phosphoinositide 3-Kinase δ Syndrome: The European Society for Immunodeficiencies-Activated Phosphoinositide 3-Kinase δ Syndrome Registry. Front Immunol. 2018;9:543. doi: 10.3389/fimmu.2018.00543.
- Coulter TI, et al. Clinical spectrum and features of activated phosphoinositide 3-kinase δ syndrome: A large patient cohort study. J Allergy Clin Immunol. 2017;139(2):597-606.e4. doi: 10.1016/j.jaci.2016.06.021.
Funding
This project is funded by a collaborative grant from Pharming.
The effect of monogenic CTLA4 mutations to human immune homeostasis
Cytotoxic T lymphocyte antigen 4 (CTLA-4) is an essential negative immune regulator that is constitutively expressed on regulatory T cells and upregulated on activated T cells. CTLA-4 inhibits T cell function by outcompeting CD28 for its binding to the co-stimulatory molecules CD80 and CD86, which are expressed on antigen-presenting cells (APCs). After binding, CTLA-4 removes CD80/CD86 from the surface of APCs and internalises it by a mechanism called transendocytosis. Thus, CTLA-4 plays an important role in maintaining peripheral tolerance and controlling T cell-driven immune response.
Heterozygous germline mutations in human CTLA4 result in an autosomal dominant immune dysregulation syndrome with incomplete penetrance known as CTLA-4 insufficiency. It is characterised by hypogammaglobulinemia, recurrent infectious diseases, various autoimmune diseases, and lymphocytic infiltration into multiple organs. Variants in CTLA4 are frequently identified in immune dysregulation cohorts, however, the functional relevance of each variant needs to be determined before a diagnosis can be made.
We have established a transendocytosis assay that allows us to measure the impact of CTLA4 variants on the ability of CTLA-4 to engulf CD80 and CD86 ligands. Using this assay, we have evaluated 35 CTLA4 variant carriers so far, and are currently analysing more.
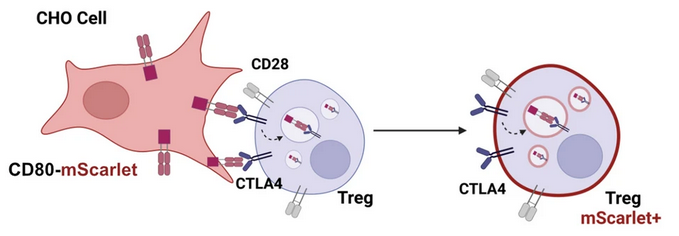
Rojas-Restrepo et al., J Clin Immunol., 2023
Interestingly, not all individuals carrying variants develop symptoms, indicating an incomplete disease penetrance of approximately 70 %. Moreover, the clinical presentation and organ involvement vary significantly among those affected. These findings suggest the existence of modifiers that either trigger disease onset or influence its severity. While several potential modifiers (such as HLA or past infections) have been investigated as potential disease modifiers in the context of CTLA-4 insufficiency, none have been conclusively identified thus far. Proposed modifiers include a gain-of-function mutation in JAK3 and a loss-of-function mutation in CLEC7A. However, these rare variants have only been observed in individual patients.
In our pursuit to identify modifiers for CTLA-4 insufficiency, we aim to test the hypothesis that affected CTLA4 mutation carriers exhibit abnormal molecular signatures. These signatures can potentially be discerned through a combination of epigenome, transcriptome, and/or proteomic profiling. Furthermore, after excluding a second somatic hit in CTLA4 itself and germline mutations in other genes within the TCR signaling pathway, we are expanding our search for genetic modifiers of disease penetrance by investigating somatic hits in the whole exome and exploring differences in the genetic makeup between affected and unaffected mutation carriers. Additionally, we are examining whether specific TCR sequences are over-represented in affected patients and whether these sequences can be linked to a specific set of viral or autoantigens.
Finally, we are investigating the potential role of the microbiome as a modifier of disease. The microbiome is known to play a critical part in shaping immune responses and maintaining tolerance to self-antigens. Our group has studied the composition of the gut microbiota in patients with common variable immunodeficiency (CVID), a disease that many CTLA4 mutation carriers suffer from. Additionally, autoimmune gut disease is commonly observed in CVID and is one of the most common organ involvements in CTLA-4 insufficiency. We have found that CVID patients have an altered gut microbiota composition characterized by decreased alpha diversity and the expansion of the class Gammaproteobacteria. Based on these findings, we aim to explore whether the gut microbiota acts as a disease modifier in CTLA-4 insufficiency.
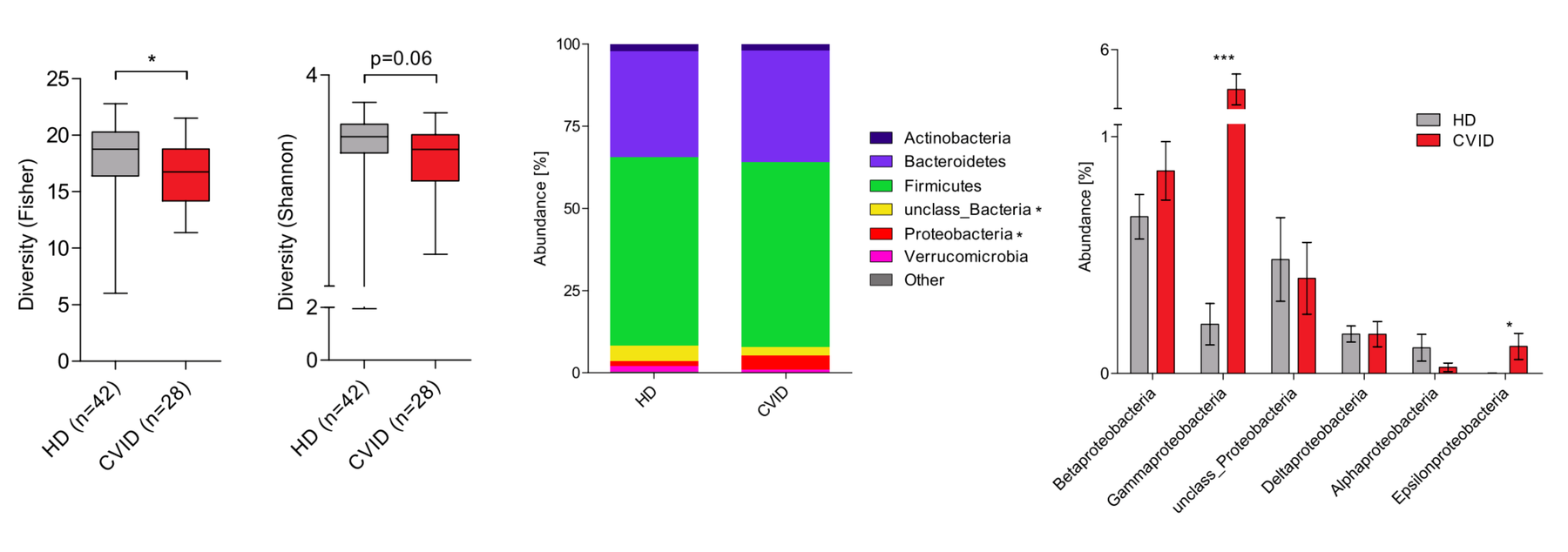
Modified from Nöltner et al, J. Clin. Immunol. (2023) doi:10.1007/s10875-023-01469-9
The discovery of modifiers for CTLA-4 insufficiency shows potential for enhancing patient care, enabling early intervention, refining prognosis, and advancing personalized medicine.
References
- Rojas-Restrepo J, et al. Functional Relevance of CTLA4 Variants: an Upgraded Approach to Assess CTLA4-Dependent Transendocytosis by Flow Cytometry. J Clin Immunol. 2023;43(8):2076-89. doi: 10.1007/s10875-023-01582-9.
- Krausz M, et al. Do common infections trigger disease-onset or -severity in CTLA-4 insufficiency? Front Immunol. 2022;13:1011646. doi: 10.3389/fimmu.2022.1011646.
- Krausz M, et al. The ABACHAI clinical trial protocol: Safety and efficacy of abatacept (s.c.) in patients with CTLA-4 insufficiency or LRBA deficiency: A non controlled phase 2 clinical trial. Contemp Clin Trials Commun. 2022;30:101008. doi: 10.1016/j.conctc.2022.101008.
- Rojas-Restrepo J, et al. Establishing the Molecular Diagnoses in a Cohort of 291 Patients With Predominantly Antibody Deficiency by Targeted Next-Generation Sequencing: Experience From a Monocentric Study. Front Immunol. 2021;12:786516. doi: 10.3389/fimmu.2021.786516.
- Egg D, et al. Therapeutic options for CTLA-4 insufficiency. J Allergy Clin Immunol. 2022;149(2):736-46. doi: 10.1016/j.jaci.2021.04.039.
- Egg D, et al. Increased Risk for Malignancies in 131 Affected CTLA4 Mutation Carriers. Front Immunol. 2018;9:2012. doi: 10.3389/fimmu.2018.02012.
- Schwab C, et al. Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4-insufficient subjects. J Allergy Clin Immunol. 2018;142(6):1932-46. doi: 10.1016/j.jaci.2018.02.055.
- Schubert D, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med. 2014;20(12):1410-6. doi: 10.1038/nm.3746.
Funding
The search for genetic modifiers is funded by the German Federal Ministry of Education and Research (BMBF) through a grant to the German Auto-Immunity Network (GAIN), grant code 01GM2206A; the search for non-genetic modifiers receives support by the Deutsche Forschungsgemeinschaft (DFG) as part of the Freiburg research consortium IMPATH under grant code SFB1160/2_B5. In addition, this work was supported in part by the Center for Chronic Immunodeficiency (CCI), Freiburg Center for Rare Diseases (FZSE). Some samples have been taken from the CCI-biobank, a partner of the Freeze Biobank Freiburg.
LRBA regulates the availability of CTLA4, autophagy, and cell metabolism
In 2012 we identified LRBA as being mutated in patients with autosomal recessive immune dysregulation and immunodeficiency (Lopez-Herrera et al., Am J Hum Genet, 2012). Following this discovery, we found that LRBA regulates autophagy. Others (Lo et al.; Science, 2015) identified LRBA as a key regulator of CTLA-4 trafficking. This project now evaluates the role of LRBA in cancer, its role in cell metabolism, and how we can use this knowledge for human diseases. This project is funded by the Wilhelm Sander-Stiftung, Förderantrags-Nr.2023.115.1.
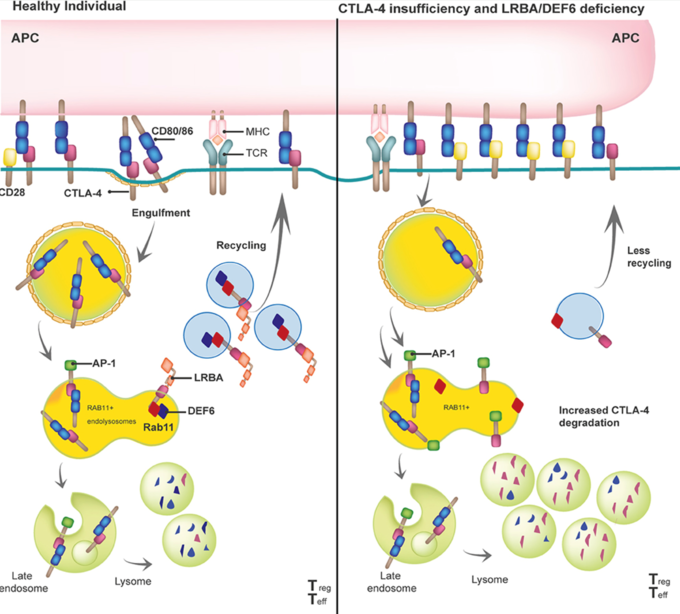
By binding to the intracellular tail of CTLA-4, LRBA protects CTLA-4 from being degraded in the lysosomal compartment and helps shuttling CTLA-4 back to the surface of e.g. regulatory T cells. In patients with biallelic pathogenic mutations in LRBA, this process is disturbed, less CTLA-4 ends up on the surface of regulatory T cells, and the clinical picture of a so-called T-reg-opathy (immune dysregulation due to an activated T cell compartment) develops. Taken from: Gámez-Díaz and Seidel, Front Pediatr, 2021.
References
- Gámez-Díaz L, Seidel MG. Different Apples, Same Tree: Visualizing Current Biological and Clinical Insights into CTLA-4 Insufficiency and LRBA and DEF6 Deficiencies. Front Pediatr. 2021;9:662645. doi: 10.3389/fped.2021.662645.
- Gámez-Díaz L, et al. The extended phenotype of LPS-responsive beige-like anchor protein (LRBA) deficiency. J Allergy Clin Immunol. 2016;137(1):223-30. doi: 10.1016/j.jaci.2015.09.025.
- Lopez-Herrera G, et al. Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet. 2012;90(6):986-1001. doi: 10.1016/j.ajhg.2012.04.015.
Applying novel technologies to the cerebrospinal fluid and its cells for the benefit of improved diagnosis and treatment prediction in neuro-immunological diseases and cerebral infections
The cerebrospinal fluid (CSF) contains soluble molecules such as protein, sugar and lipids, and – at least in the healthy situation – only few cellular components, i.e up to 3 cells per microliter. The analysis of CSF could be performed by many different methods, such as fluorescent-activated-cell-sorting (FACS), single cell transcriptomics (scRNAseq), or mass-spectrometry (MS/MS). However, in clinical practice only a differential blood cell count, some limited clinical chemistry measures, and immunoglobulin analysis is routinely being used (1). This routine analysis serves many purposes, but has its limitation in e.g. differentiating a viral CNS infection from an autoimmune CNS process.
In our CSF study, we analyze and compare the following methods, for an improved characterization of the content of the CSF, and hence the benefit of our patients:
- The cell type characterization by flow cytometry (FACS)
- A novel epigenetic analysis of the cellular components of the CSF (developed by the company EPIMUNE) will be compared to
- As a gold standard, single-cell RNA sequencing for the comprehensive analysis of cells in the CSF will be performed in selected samples.
In collaboration with the Departments of Neurology (Prof. S. Rauer), Psychiatry (Prof. L. Tebartz van Elst), and Gastroenterology (Dr. Sagar) we are examining the CSF of patients with a broad variety of neurological diseases, including but not limited to viral and bacterial infections and several autoimmune conditions. We postulate that different cell types can be measured when the total cell count in the CSF is high. We suspect that in certain patient groups, numerical differences in cell types will emerge. The CSF of these patients is spun down into a cellular pellet, which are then being analyzed by the following methods:
1. Flow cytometry
A routinely-used method to count difference cell subsets in blood is flow cytometry. Despite its challenge to detect low numbers of cells in samples, we have developed a methodological approach to overcome these problems: Utilizing the advanced capabilities of the Sony ID7000 Spectral Cell Analyzer allows for a precise interrogation of an antibody panel consisting of 21 different antibodies in a single sample. This strategic selection enables comprehensive phenotypic profiling of different leukocyte subsets beyond the traditional scope of analysis, ensuring optimal resolution and accuracy in our analytical assessments.
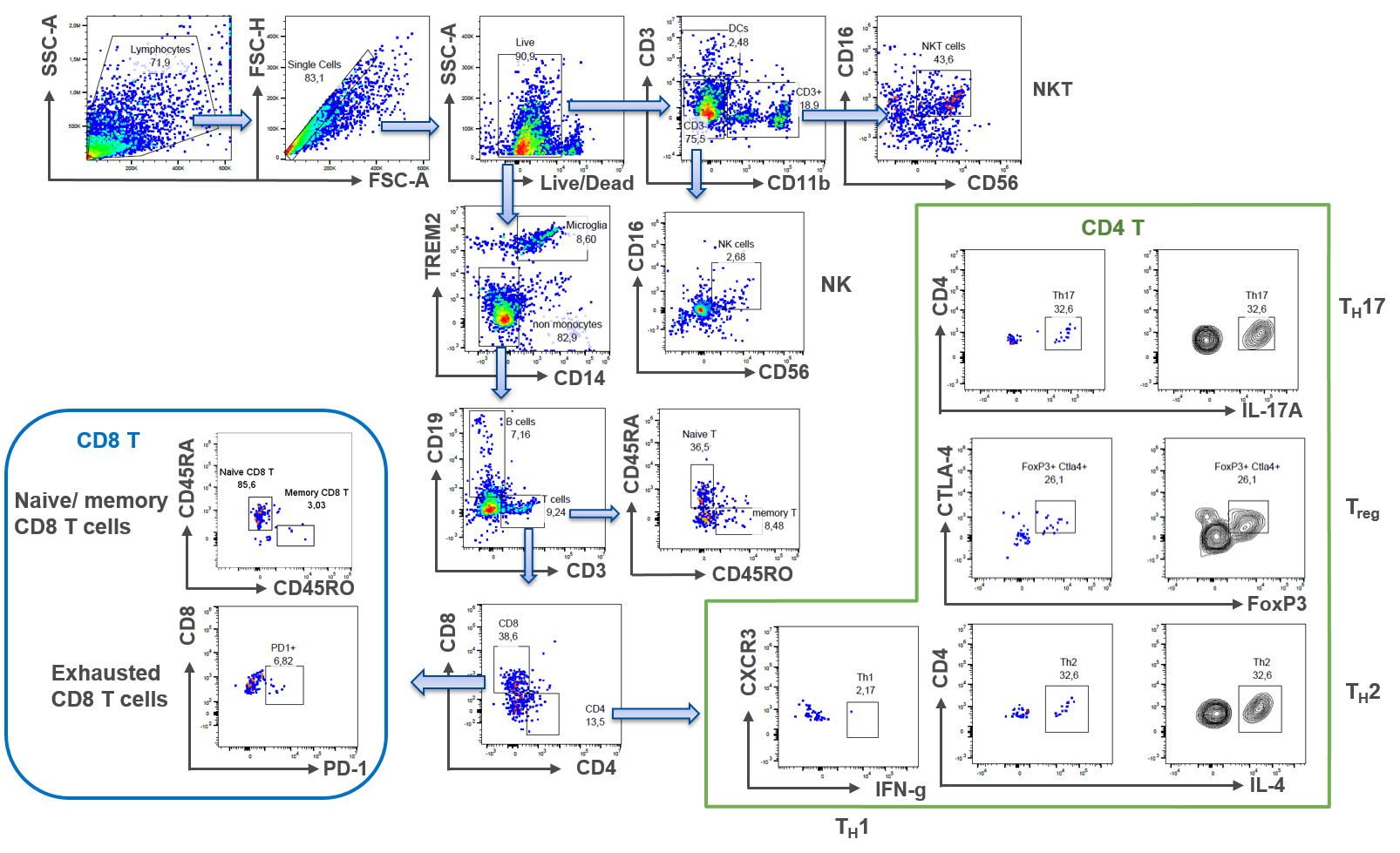
2. Epigenetics
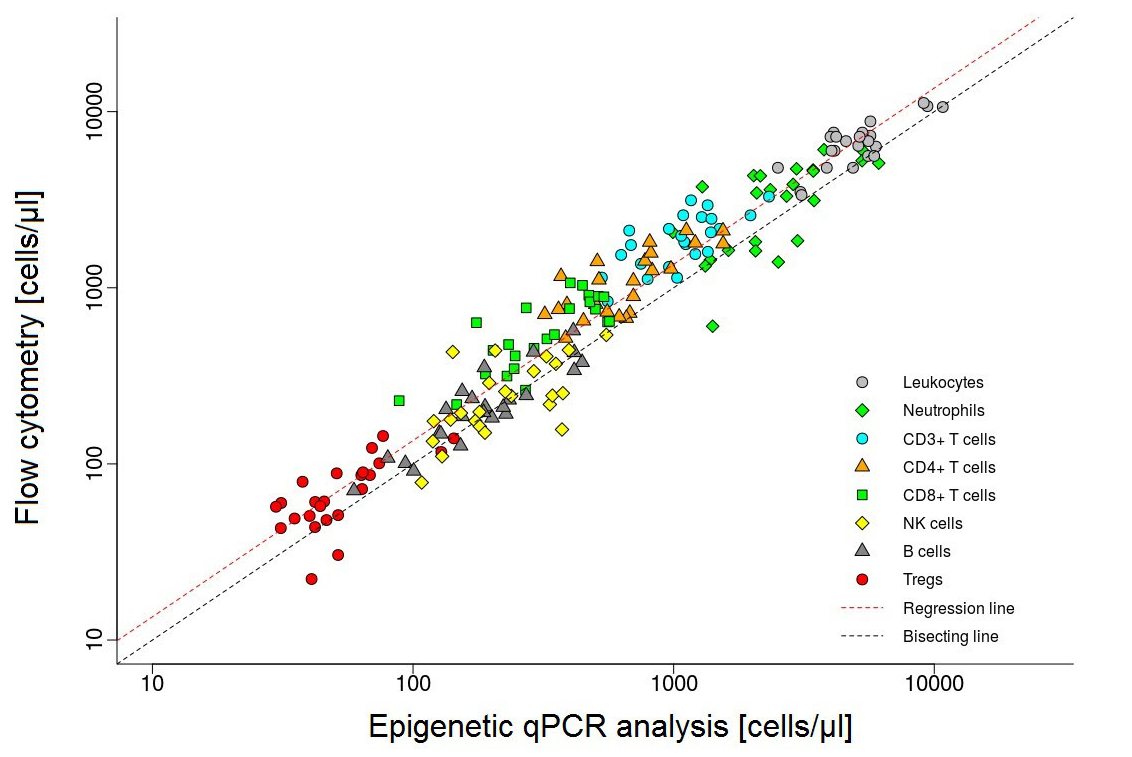
Different cell types can be distinguished by their DNA methylation signature. DNA methylation, and hence the epigenetic signature of cells can be examined by a quantitative PCR (qPCR). This method has been developed by a company (EPIMUNE GmbH, Berlin) for the peripheral blood. In blood, the percentual quantity of the leukocyte subsets of CD3-, CD4-, CD8-, TH17-, Treg-cells, PD-1 molecules, B-cells, NK-cells, as well as monocytes and neutrophils, can be detected and compared. We would like to run this method on cerebrospinal fluid to determine the exact origin and capability of cells within the CSF.
3. Single cell RNA sequencing – the gold-standard
We will also examine some selected samples by single-cell RNA sequencing. This method is considered to be very reliable but requires a lot of effort to be performed. Above that, the costs are quite high. Nevertheless, this method is currently considered our gold-standard and is essential for the comparison of the epigenetic- and the flow cytometry-based approaches.
Reference
- Otto F, et al. Role and Relevance of Cerebrospinal Fluid Cells in Diagnostics and Research: State-of-the-Art and Underutilized Opportunities. Diagnostics (Basel). 2021;12(1) doi: 10.3390/diagnostics12010079.
Funding
This work is supported by the Center for Chronic Immunodeficiency (CCI), Freiburg Center for Rare Diseases (FZSE).
What are multi-organ autoimmune diseases?
Multi-organ autoimmune diseases belong to the “ultra-rare” disorders. In this case, the body’s own immune system mistakenly attacks its own organs. Affected individuals then develop inflammation of several organs, for example the bone marrow, intestines, lungs, kidneys, skin and nervous system. Due to the rarity and complexity of the disease patterns, it often takes a long time before the correct diagnosis is made.
Single genes and monogenetic mutations have already been discovered as the cause of this group of diseases. These and other findings also help to improve the understanding and treatment for more common polygenic autoimmune diseases.

Joint research in the GAIN network
In the GAIN research network, experts at various German locations are working together to research the causes and therapies of genetic multi-organ autoimmune diseases. The Federal Ministry of Education and Research (BMBF) has been funding the network since 2019.
In order to facilitate diagnosis and counseling of affected individuals and their families and to improve treatment, the individual research teams are looking into the underlying molecular and cellular pathomechanisms of individual diseases as well as possible molecular interventions as a therapeutic option. In this context, already known but also new genetic causes of disease are analyzed comprehensively. Samples from a biomaterial bank are already available to the researchers for their work. Another focus is on the development of a uniform approach to the identification, diagnosis and treatment of multi-organ autoimmune diseases.
Available data are systematically collected in a patient registry. Through this registry, patients could be recruited for a companion clinical trial investigating the safety and efficacy of the immune-modulatory drug Abatacept in a first funding period (2019-2022). In the second funding period (2023-2025), the quality of life of patients will now be investigated with questionnaires and patient involvement in order to better address their everyday problems.
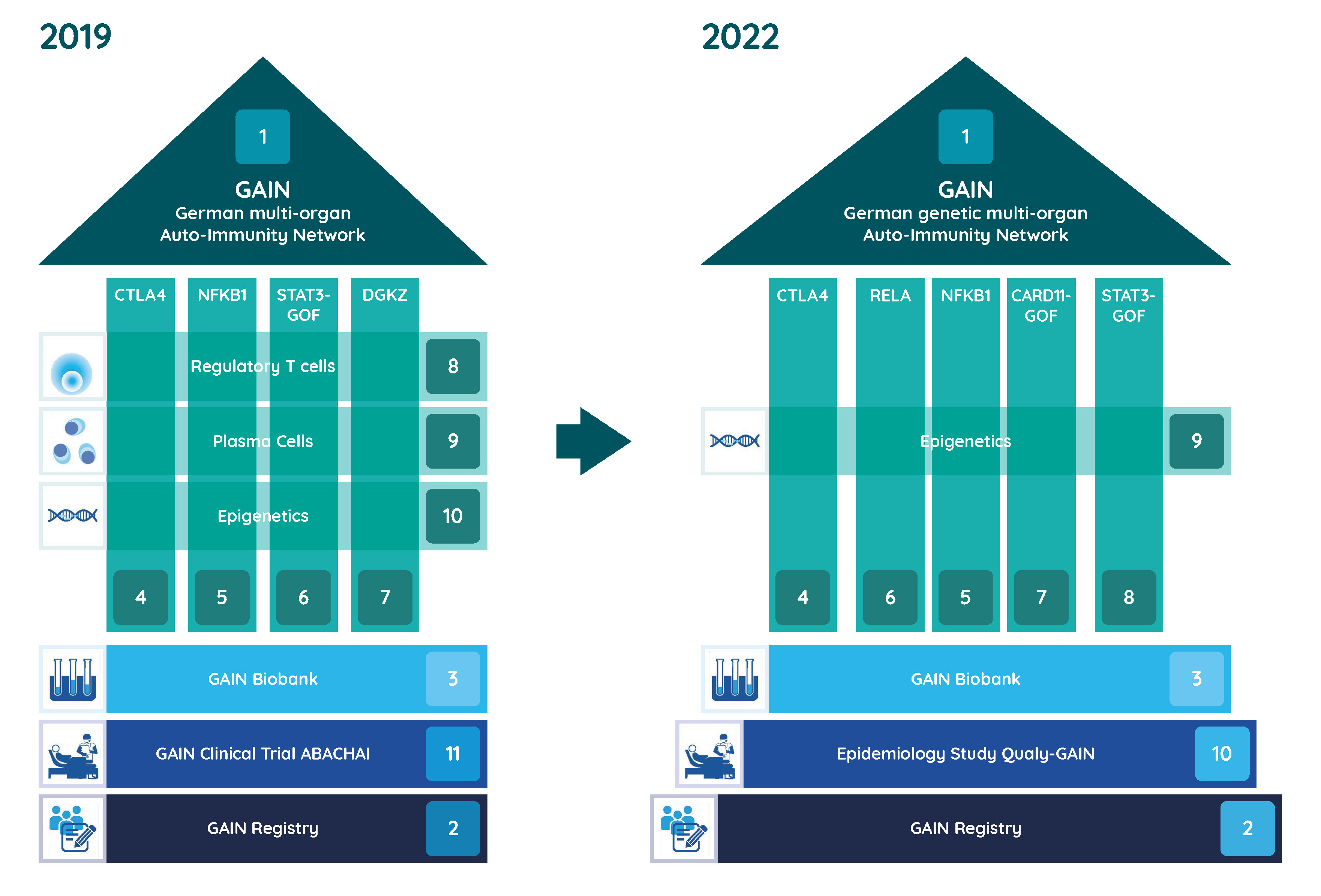
Projects
- Coordination of GAIN, Prof. Dr. med. Bodo Grimbacher, Freiburg
- Registry of the German genetic multi-organ Auto-Immunity Network (GAIN-registry), Prof. Dr. med. Ulrich Baumann, Hanover, Dr. med. Dipl. Inf. Gerhard Kindle, Freiburg, Prof. Dr. rer. nat. Alexandra Nieters, Freiburg
- Consortial Biobank for patients with Inborn Errors of Multi-Organ Autoimmune Diseases, Prof. Dr. rer. nat. Thomas Illig, Hanover
- CTLA4 insufficiency, Prof. Dr. med. Bodo Grimbacher, Freiburg
- Immune dysregulation due to NFKB1D defects, Prof. Dr. med. Klaus Warnatz, Freiburg
- Type I IFN-driven autoinflammation and autoimmunity caused by heterozygous truncating mutations in RELA, Prof. Dr. Min Ae Lee-Kirsch, Dresden
- The role of activating mutations in CARD11 on the immune system, Prof. Dr. Dr. med. Fabian Hauck, Munich
- STAT3 gain-of-function (GOF) associated disease, Prof. Dr. med. Stephan Ehl, Freiburg
- Identification of epigenetic factors in multi-organ autoimmunity, Dr. rer. nat. Faranaz Atschekzei, Hanover, Prof. Dr. Torsten Witte, Hanover
- Qualy-GAIN – an epidemiological study on the quality of life of GAIN patients, Prof. Dr. Erik Farin-Glattacker, Freiburg, Prof. Dr. med. Jochen Schmitt, Dresden
Projects 2019 - 2022
- Initial description of human DGKζ -deficiency, Prof. Dr. Dr. med. Fabian Hauck, Munich
- The role of GARP in monogenic traits of multi-organ autoimmunity, Prof. Dr. Alla Skapenko, Munich, Prof. Dr. med. Hendrik Schulze-Koops, Munich
- Monogenetic immune dysregulation syndromes and their effect on the plasma cell compartment, Prof. Dr. med. Bimba Franziska Hoyer, Kiel, Prof. Dr. rer. nat. Andreas Radbruch, Berlin
- Safety and Efficacy of abatacept (s.c.) in patients with CTLA4 insufficiency and LRBA deficiency (ABACHAI), Prof. Dr. med. Bodo Grimbacher, Freiburg
Publications
CV
| Since 2025 | Scientific Director, CCI, Medical Center - University of Freiburg |
| 2021-2022 | Sabbatical at the University of California San Diego (UCSD), USA |
| Since 2019 | Vice Director of the Institute for Immunodeficiency (IFI) at the CCI, Medical Center – University of Freiburg |
| 2011-2019 | Scientific Director and Consultant, CCI, Medical Center - University of Freiburg |
| 2006-2011 | Consultant and EU Marie-Curie Research Group Leader, Department of Immunology, Royal Free Hospital, University College London, UK |
| 2006 | Habilitation in Internal Medicine, University of Freiburg (Prof. Dr. Hans-Hartmut Peter) |
| 2000-2006 | Physician, Department of Rheumatology and Clinical Immunology, Medical Center - University of Freiburg |
| 1997-2000 | Postdoc, National Human Genome Research Institute (NIH), Bethesda, Maryland, USA |
| 1995-1997 | Assistant Physician, Department of Rheumatology and Clinical Immunology, Medical Center - University of Freiburg |
| 1995 | Dissertation in Medicine, University of Freiburg (Prof. Dr. Hermann Eibel) |
| 1988-1995 | Study of Medicine in Aachen, Freiburg, and Hamburg |
Team


Prof. Dr. med. Bodo Grimbacher
+49 (0)761 270-77731
+49 (0)761 270-77744
Center for Chronic Immunodeficiency
at Center for Translational Cell Research
Breisacher Str. 115
79106 Freiburg
Germany