Selected Projects in Pillar 1: Therapeutic Innovations
The projects selected under Therapeutic Innovations address key challenges in precision oncology and translational therapy development:
Treatment failure and tumour recurrence due to survival of therapy-resistant prostate cancer (PCa) cells in patients represent major clinical issues to overcome and is a still highly unmet medical need. Our lab identified and validated lysine methyltransferase 9 (KMT9) as a novel target for the treatment of castration-resistant prostate cancer (CRPC). KMT9 is a heterodimer composed of KMT9α and KMT9β that monomethylates histone H4 at lysine 12 (H4K12me1) in PCa and other tumours such as bladder, colon and lung. KMT9 controls transcription of target genes involved in cell cycle regulation thereby promoting tumour cell proliferation. Importantly, KMT9 loss or enzymatic inactivation blocks proliferation of prostate cancer cells and organoids in vitro, in xenografted tumours, and in genetically engineered prostate tumour bearing mice. Thus, the KMT9 mechanism of action provides a promising novel therapeutic paradigm for the treatment of CRPC exceeding the current gold standard of care. In consequence, we developed highly potent and selective KMT9 inhibitors that impair proliferation of prostate tumour cells. Our KMT9 inhibitors are extremely potent and selective in rodent and human cells and tissues and show very favourable pharmacokinetic properties in vivo in mice and rat. In the DKTK level 2 funded project, we aim to generate for the two most promising drug candidates a preclinical data package that will enable us to move them forward to investigational new drug (IND) filling and clinical phase I testing. Thus, in this project we plan to profile the efficacy and selectivity of our selected drug candidates in vitro and in vivo.
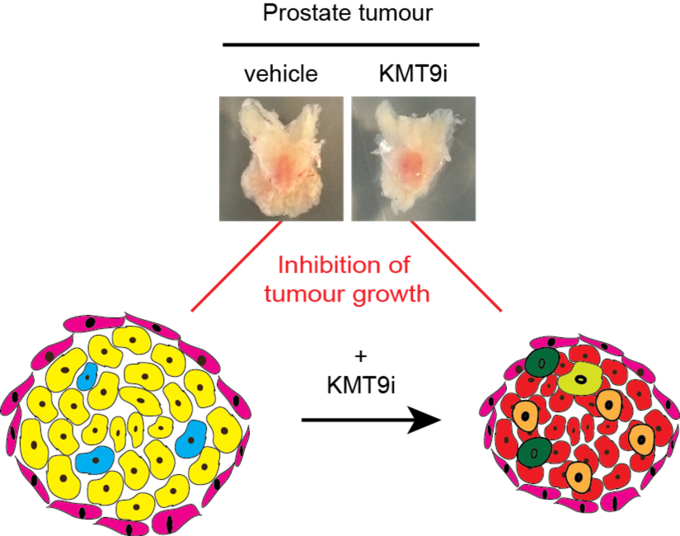
Treatment with KMT9 inhibitor (KMT9i) blocks growth of prostate tumour in mice.

Dr. Eric Metzger
Molecularly targeted cancer therapy faces two major challenges: First, genetic variants of uncertain significance (VUS), whose functional impact and druggability remains unknown, are not informative for clinical decisions in molecular tumor boards (MTBs). Second, the targeted monotherapy leads to the development of secondary resistance requiring rationally designed combinatorial therapies.
Oncogenic FGFR aberrations drive tumorigenesis in many tumor entities. FGFR inhibitors (FGFRis) have been approved by the FDA/EMA, but tumors exhibit either primary resistance or acquire secondary resistance after an initial response, often in less than a year. FGFRi therapies are approved for FGFR translocations, while the numerous point mutations and common amplifications are largely of uncertain significance and thus not routinely treated with FGFRis.
The “hitFGFR” project aims to tackle both these major challenges to target FGFR-driven tumors more effectively in patients. Previously, we identified hundreds of clinically relevant activating and resistance-mediating FGFR mutations in the kinase domains of FGFR1 - FGFR4by saturation mutational scanning (Tangermann et al., Nature Genetics 2026). Clearly, FGFRi monotherapy will not eradicate the disease in a significant number of patients, thus requiring combinatorial approaches including different kinase inhibitors and/or immune checkpoint blockade (ICB).
HitFGFR will classify all possible FGFR point mutations for their activation and drug resistance based on robust functional data and thus make them practically exploitable for clinical decisions, identify off-target resistance mechanisms to design combinatorial treatment approaches which prevent/overcome tumor-intrinsic FGFRi resistance, and explore the in vivo synergy of ICB and FGFRi in a murine lung cancer model. Importantly, the results will be directly translated to treatments in molecular tumor boards and shall contribute to the preparation of a prospective interventional clinical trial.
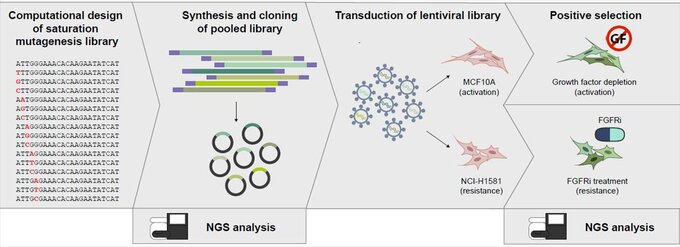
For Saturation Mutational Scanning, every single nucleotide in the Fibroblast Growth Factor Receptor genes FGFR1-FGFR4 is mutated to every other nucleotide generating a library of all possible 29259 point mutations. These libraries are then lentivirally transduced into two different model systems for positive selection screens quantifying the impact of each mutation on activation and drug response.

Prof. Dr. Sven Diederichs
CAR-NK Cell Therapy in Juvenile Myelomonocytic Leukemia
Background and Unmet Need
Juvenile myelomonocytic leukemia (JMML) is a malignant myeloproliferative disorder affecting young children. Allogeneic hematopoietic stem cell transplantation (HSCT) currently represents the only curative option, yet the 5-year event-free survival does not exceed 60%, underscoring the need for innovation.
Therapeutic Target: CLEC12A
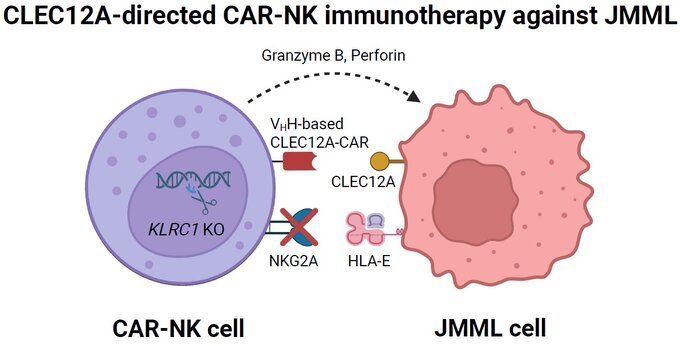
CLEC12A is a leukemic stem cell antigen highly expressed on JMML-initiating cells while being absent on normal human hematopoietic stem cells. This selectivity offers a unique therapeutic window: leukemic cells can be targeted without compromising healthy hematopoiesis, making CLEC12A a compelling and biologically rational target in this disease.
Why CAR-NK Rather Than CAR-T?
Chimeric antigen receptor (CAR) T-cell therapy has transformed treatment of refractory or relapsed lymphoid malignancies, but has yet to be translated to myeloid diseases including JMML. This project develops allogeneic CAR natural killer (NK) cell preparations instead, for two key reasons. First, CAR-T cells must be manufactured from autologous blood, which is frequently not feasible given the compromised clinical condition of JMML patients at diagnosis. Second, NK cells do not induce graft-versus-host disease and are associated with a substantially more favorable safety profile.
Project Design and Innovation
The project brings together the complementary expertise of two project partners to evaluate CLEC12A-directed CAR-NK cells in established preclinical JMML models. Four engineering innovations converge in the final cell product:
A second-generation CLEC12A-targeting CAR incorporating a 4-1BB co-stimulatory domain has already been constructed and optimized. Highly affine and specific variable domains of heavy chain-only antibodies (VHH, "nanobodies") have been isolated to drive target recognition. An IL-15 transgene is co-inserted into the NK cells to support their in vivo proliferation and persistence. Finally, CRISPR-Cas9-mediated knockout of KLRC1 eliminates the inhibitory checkpoint receptor NKG2A, providing a further functional enhancement of the final product.
Bridge to Clinical Translation
Following preclinical efficacy evaluation, the CAR-NK cells can be transferred directly to clinical-grade production using existing GMP-certified manufacturing infrastructure. An additional combinatorial strategy is supported by prior observations that azacitidine increases CLEC12A surface expression, thereby enhancing NK cell-mediated cytotoxicity — an approach already validated in AML that will inform the clinical design here. Clinical evaluation will be conducted within the multinational framework of the European Working Group of Myelodysplastic Syndromes in Childhood (EWOG-MDS), headquartered and coordinated at the DKTK partner site in Freiburg.

Prof. Dr. Christian Flotho